Avant-propos
Items fantômes est une fructueuse opération de procrastination.
D'abord par son contenu. Il est fort peu probable que des chapitre de ce livre fasse l'objet de questions discriminantes au concours de l'internat (du moins je l'espère). Sa lecture a donc peu de chances d'être rentable (mon agent m'a conseillé de ne pas mettre en avant ce point dès l'avant-propos, mais je ne l'ai pas écouté).
Également par la chronologie de sa rédaction. De nombreux chapitres ont été écrits en cours de 6e année sur du temps libre qui aurait été mieux employé à réviser les ECOS.
Enfin par le temps déraisonnablement passé sur de ridicules détails. Je préfère ne pas dire combien de temps j'ai passé à peaufiner l'apparence des chapitres ou bricoler avec le code sous-jacent à ce site internet.
On pourrait également ajouter la procrastination à finalement publier un ouvrage dont le contenu est bouclé depuis plusieurs mois... Pour ma défense, je voulais à tout prix éviter de le libérer dans la nature à l'approche des EDN pour ne pas créer une FOMO d'une lecture somme toute fort dispensable.
Procrastination donc, mais procrastination utile toutefois. Utile pour se changer les idées dans une 6e année pleine de désillusions. Utile pour patienter jusqu'aux résultats des ECOS. Et peut-être utile pour vous qui lirez ce livre.
Je tiens à remercier l'électricité sans qui rien de tout cela ne serait possible.
En espérant que l'exploration des items fantômes vous apporte quelques frissons médicaux, bonne lecture !
Remerciements
Ce livre n'aurait pas eu la structure qu'il a aujourd'hui sans les perspicaces suggestions d'Abderrahmane Bousta qui ont grandement contribué à la rigueur et la standardisation des différents chapitres.
Réseau ferré des items fantômes
Introduction
Items fantômes a pour objectif de présenter des pathologies qui apparaissent à de multiples reprises dans les manuels de médecine qui préparent au concours de l'internat sans pour autant figurer en tant que tel au programme de ce concours.
Pourquoi ?
L'idée de cet ouvrage a fait son bout de chemin en partant de plusieurs constats.
Tout d'abord l'existence de plusieurs pathologies qui cochent les cases de cette définition. On a tous lu des dizaines de fois "syndrome de Gougerot-Sjögren", "lichen plan", ou "cholangite biliaire primitive" sans avoir la moindre idée de ce dont il s'agit et sans jamais trouver de chapitres dédiés dans nos manuels.
Ces maladies, à la fois là et pas là, restent souvent des boîtes noires pour des étudiant·e·s qui ne peuvent prendre la peine de les ouvrir faute de sources simplement accessibles à leur sujet et surtout faute de temps. Bien qu'omniprésentes, elles deviennent des items fantômes.
Mais connaître un nom de maladie sans savoir quoi mettre derrière peut être particulièrement frustrant et participe au sentiment d'inanité voire d'absurdité de nos études : à quoi bon investir son temps et son énergie pour des connaissances superficielles et inexploitables ?
C'est là qu'intervient Items fantômes qui a été conçu pour apporter des solutions à ces constats : un livre qui aborde succinctement ces maladies ectoplasmiques pour y lever le voile.
Comment ?
Cette première version se veut synthétique et accessible. La même ossature est utilisée pour tous les chapitres :
- un (très) bref résumé de la pathologie en question selon un plan stéréotypé ;
- une liste des items du programme où elle est abordée ;
- un renvoi vers quelques ressources pour approfondir le sujet (ce sont ces ressources qui nous ont servi de base pour rédiger les chapitres).
Les chapitres ont été écrit par externes et des internes, avec toutes les réserves que cela implique sur la qualité des informations que vous trouverez ici. Si vous identifiez des erreurs (il y en a sûrement pas mal), n'hésitez pas à nous contacter.
À noter
Le sommaire que nous avons constitué n'est assurément pas exhaustif. Si vous identifiez d'autres items fantômes, n'hésitez pas à nous contacter pour que nous puissions les y intégrer.
En attendant, plusieurs sources s'offrent généralement à vous :
- s'il s'agit d'une maladie rare, il y a fort à parier qu'elle bénéficie d'un Plan National de Diagnostic et de Soins. Ce sont de précieux documents qui fournissent une excellent introduction à ces pathologies ;
- les énormes traités de médecine, souvent au rayon WB de vos BU, abordent à peu près toute la médecine ;
- Google peut toujours faire l'affaire, a fortiori quand on traduit les mots en anglais.
Licence & Crédits
Pour le contenu dont nous ne disposons pas des droits, la licence de partage est indiquée en passant son curseur sur la sourceCeci est un easter egg.. Il s'agit principalement des illustrations.
Pour tout le reste et quand c'est applicable, le livre est distribué sous la licence CC BY-NC-SA 4.0.
Si du contenu dont vous détenez les droits se trouve dans cet ouvrage et qu'il n'est pas attribué de la bonne manière (ou que vous souhaitez le voir retirer), n'hésitez pas à nous contacter pour que l'on règle le soucis aussi vite que possible.
Ce livre a été créé à l'aide de mdbook.
L'auteur
Léo Picat était externe quand il a lancé ce projet et pas techniquement interne quand sa première version fut achevée.
Contact
leo[point]n[point]picat[arobase]gmail[point]com
Mode d'emploi
Comment utiliser Items fantômes ?
De par ses objectifs, le contenu d'Items fantômes n'est bien entendu pas à apprendre par cœur. Deux usages me semblent les plus évidents :
- un dictionnaire : quand vous rencontrez ce que vous pensez être un item fantôme, ouvrez Items fantômes et regardez si votre suspect figure au sommaire de l'ouvrage ;
- une conclusion d'item : une fois que vous avez fini de travailler un item,
cherchez
Item \son-numérodans la barre de recherche (auquel on accède via la loupe en haut de la page). Cela affichera toutes les pages qui mentionnent l'item.
Le livre étant entièrement gratuit, vous en faites bien ce que vous voulez bien évidemment.
Plan
Le plan de tous les chapitres est le même.
Vous trouverez donc :
- les éléments essentiels sur la maladie à commencer par un résumé en une phrase
puis :
- des éléments épidémiologiques sur la maladie, généralement incidence/prévalence, âge moyen, et sex ratio ;
- les signes de la maladie ;
- les causes de la maladie, qu'ils s'agissent des mécanismes physiopathologiques à l'origine des signes ou la liste des étiologies ;
- les éléments nécessaires au diagnostic de la maladie, qu'ils soient positifs, étiologiques, ou de gravité ;
- de très brefs éléments sur le traitement ;
- parfois quelques illustrations quand elles ont un intérêt en lien avec le programme ou pour faciliter la lecture du reste du chapitre.
- la liste des items dans lesquelles la maladie est abordée et pourquoi elle y est mentionnée ;
- des références vers lesquelles se tourner pour approfondir le sujet.
Les références
Une source qui nous a été très utile dans la rédaction est UpToDate, une site de recommandation américain extrêmement sourcé et qui semble couvrir toute la médecine. Des astuces pour y accéder sont présentes dans les bonus.
Si jamais des liens sont cassés, ce qui arrivera très probablement si la HAS
modifie son site, vous pouvez tenter une recherche Google \nom-de-la-maladie \nom-du-lien. Vous pouvez aussi tenter votre chance sur WayBack Machine : lorsque
cela nous semblait pertinent, nous y avons sauvegardé les liens sortants de cet
ouvrage.
Thème
Le petit pinceau en haut de la page vous permet de changer le thème du livre. Le
thème Rust est celui qui offre les meilleurs contrastes mais qui rappelle
aussi beaucoup les années 70...
Me soutenir
Si le projet vous plaît, que vous avez apprécié la lecture ou que vous trouvez ça si nul que vous voulez tout faire pour que j'arrête de produire du contenu, vous pouvez me laisser un pourboire sur cette plate-forme. L'argent servira en priorité à financer l'hébergement du site, puis probablement à acheter des bonbons. Cela étant dit, l'accès à Items fantômes restera gratuit (knowledge should be free comme on dit dans la langue de Jay-Z).
Vous pouvez aussi parler du livre autour de vous pour qu'il puisse être utile à quiconque en ressentirait le besoin.

Syndrome de Gougerot Sjögren
- En (très) bref
- Items où il est abordé
- Item 90 Pathologies des glandes salivaires
- Item 96 Neuropathies périphériques
- Item 192 Pathologies auto-immunes : aspects épidémiologiques, diagnostiques et principes de traitement
- Item 261 Néphropathies glomérulaires
- Item 262 Néphropathies interstitielles chronique
- Item 267 Troubles hydro-électrolytiques
- Item 273 Dysphagie
- Item 319 Lymphomes malins
- Références
En (très) bref
Épidémiologie
Le syndrome de Gougerot-Sjögren a une prévalence comprise entre 10 et 100 pour 100 000 habitants. Il survient le plus souvent autour de 50 ans. Il touche 9 femmes pour 1 homme.
Signes
Dans sa forme classique, la maladie associe :
- un syndrome sec qui atteint l'œil, la bouche, mais aussi la peau ;
- des arthro-myalgies d'horaire inflammatoire ;
- asthénie.
Comme les autres maladies auto-immunes systémiques du programme, le syndrome de Gougerot Sjögren peut donner énormément de signes. Nous vous renvoyons à la section dédiée pour une liste partielle. Pour résumer, on peut dire que c'est une maladie des glandes exocrines.
Causes
Il peut survenir seul ou en association avec d'autres maladies auto-immunes, notamment la polyarthrite rhumatoïde, le lupus ou la sclérodermie.
Diagnostic/Examens complémentaires
Les critères de classification1 sont tous basés sur des examens complémentaires. Un score supérieur ou égal à 4 est nécessaire. Il faut également éliminer les autres causes de syndrome sec.
| Item | Points |
|---|---|
| BGSA avec sialadénite lymphocytaire | 3 |
| Anticorps anti-SSA | 3 |
| Ocular Staining Score ≥ 4 | 1 |
| Test de Schirmer ≤ 5 mm | 1 |
| Flux salivaire ≤ 0.1 mL/min | 1 |
Les anticorps associés le plus spécifiquement à la maladie sont les anticorps anti-SSA (aussi appelés anti-Ro) et anti-SSB (aussi appelés anti-La). Le facteur rhumatoïde est souvent positif également.
Traitement
La prise en charge du syndrome sec est essentielle car il impacte fortement la qualité de vie : traitement substitutif et pilocarpine sont utilisés. Les corticoïdes ne sont pas efficaces sur ce symptôme.
Les atteintes systémiques sont traitées avec des immunosuppresseurs.
Items où il est abordé
Item 90 Pathologies des glandes salivaires
C'est une cause de syndrome sec et de gonflement des glandes salivaires.
Item 96 Neuropathies périphériques
C'est une cause de neuropathie périphérique neuronale sensitive.
Item 192 Pathologies auto-immunes : aspects épidémiologiques, diagnostiques et principes de traitement
C'est l'une des maladies auto-immunes non spécifiques d'organe les plus fréquentes (plus que le lupus, tant et si bien que ce n'est forcément pas une maladie rare).
Item 261 Néphropathies glomérulaires
C'est une cause de glomérulopathie extra-membraneuse.
Item 262 Néphropathies interstitielles chronique
C'est une cause de néphropathie interstitielle chronique.
Item 267 Troubles hydro-électrolytiques
C'est une cause d'acidose tubulaire distale.
Item 273 Dysphagie
C'est une cause de dysphagie non lésionnelle par trouble moteur.
Item 319 Lymphomes malins
C'est une cause de lymphome par stimulation chronique du système immunitaire.
Références
Sclérodermie
- En (très) bref
- Items où elle est abordée
- Item 192 Pathologies auto-immunes : aspects épidémiologiques, diagnostiques et principes de traitement
- Item 210 Pneumopathie interstitielle diffuse
- Item 213 Anémie
- Item 239 Acrosyndromes
- Item 224 Hypertension artérielle de l'adulte et de l'enfant
- Item 273 Dysphagie
- Item 283 Constipation
- Item 305 Tumeurs de l'œsophage
- Références
En (très) bref
Épidémiologie
La prévalence de la sclérodermie systémique est de 15 pour 100 000 en France. Le pic de survenue est autour de 55 ans (plus ou moins 10 ans). Il y a une nette prédominance féminine avec un sex ratio oscillant entre 3 et 8 femmes pour 1 homme.
Signes
Il existe 2 formes de la maladie :
- une forme cutanée limitée où l'atteinte ne dépasse ni les coudes, ni le genoux ;
- une forme cutanée diffuse où les lésions touchent également la racine des membres voire le tronc.
Comme son nom l'indique, la sclérodermie systémique peut atteindre de nombreux organes.
Signes cutanés
La sclérose cutanée qui permet de définir les 2 formes de la maladie est quasi-systématique, bien qu'il existe des formes sine scleroderma. Elle peut être responsable d'une limitation de l'ouverture buccale.
Un phénomène de Raynaud secondaire est extrêmement fréquent. Il s'associe à des atteintes digitales à type d'ulcération, de doigts boudinés et de télangiectasie.
Signes articulaires
On peut voir des arthralgies non inflammatoires et des myalgies.
Signes digestifs
L'atteinte digestive est très fréquente même si elle n'est pas toujours parlante. On voit surtout un reflux gastro-œsophagien. On observe également une dysphagie non lésionnelle par troubles moteurs.
Signes pulmonaires
Cette atteinte peut être très grave et justifie un dépistage dès le diagnostic. On retiendra surtout :
- une pneumopathie interstitielle diffuse à type de pneumopathie interstitielle non spécifique ;
- une hypertension artérielle pulmonaire qui peut être de type 1, de type 2 en lien avec des signes cardiaques, ou de type 3 en lien avec la pneumopathie interstitielle diffuse.
Cela se traduit donc cliniquement par une dyspnée
Signes cardiaques
L'atteinte est fréquente mais souvent asymptomatique. La sclérodermie systémique peut toucher les 3 tuniques du cœur ainsi que le tissu de conduction. Elle justifie également un dépistage systématique au diagnostic.
Signes rénaux
La crise rénale sclérodermique peut concerner jusqu'à 10% des patient·e·s, principalement dans les formes cutanées diffuses. Elle associe :
- une hypertension artérielle maligne de début brutal ;
- une insuffisance rénale aiguë organique de début brutal ;
- une micrangiopathie thrombotique.
Autres signes
Chez les hommes, on trouve souvent une dysfonction érectile.
Causes
La physiopathologie de la maladie est encore mal comprise. Elle associe une activation du système immunitaire, une atteinte des vaisseaux, et une synthèse excessive de matrice extra-cellulaire.
Comme pour beaucoup de maladies auto-inflammatoires, il y a très certainement un terrain génétique prédisposant non suffisant et des facteurs environnementaux. Les gènes de prédisposition augmentent également le risque d'autres maladies auto-immunes, comme le lupus érythémateux disséminé.
Diagnostic/Examens complémentaires
Les critères de classification de la maladie combinent clinique et examens complémentaires1. Un score supérieur ou égal à 9 est nécessaire.
| Item | Points |
|---|---|
| Épaississement cutané au-delà des articulations méta-carpo-phalangiennes | 9 |
| Doigts boudinés | 2 |
| Atteintes des doigts ne dépassant pas les articulations méta-carpo-phalangiennes | 4 |
| Ulcères pulpaires digitaux | 2 |
| Cicatrices déprimées | 3 |
| Télangiectasies | 2 |
| Anomalies à la capillaroscopie | 2 |
| Hypertension artérielle pulmonaire | 2 |
| Phénomène de Raynaud | 3 |
| Anticorps anti-topoisomérase I ou anti-centromères ou anti-ARN polymérase 3 | 3 |
Les anticorps anti-topoisomérase I sont aussi appelés anti-Scl70.
Le bilan initial dépistera également les atteintes d'organes les plus graves.
Traitement
Les séances de rééducation ne sont pas à négliger, de même que les traitement symptomatiques.
Iconographie
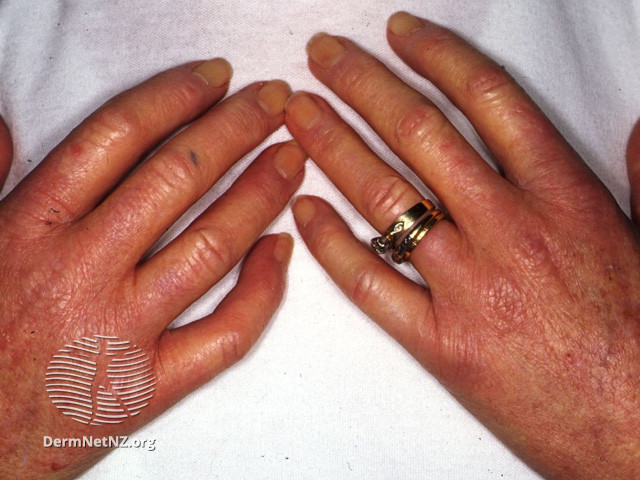
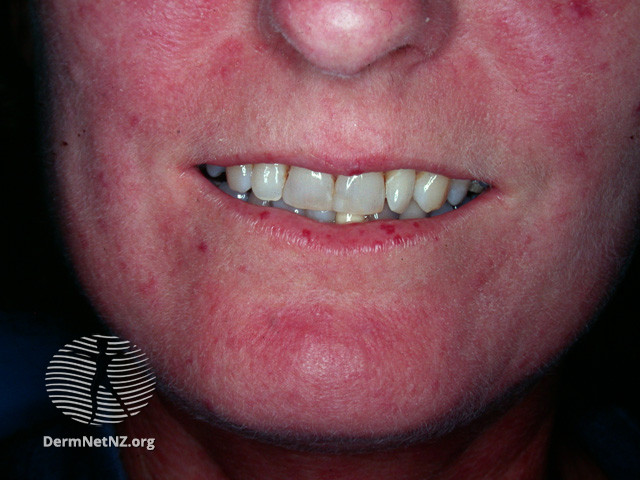
Items où elle est abordée
Item 192 Pathologies auto-immunes : aspects épidémiologiques, diagnostiques et principes de traitement
C'est une maladie auto-immune non spécifique d'organes.
Item 210 Pneumopathie interstitielle diffuse
C'est une cause de pneumopathie interstitielle diffuse, notamment à type de pneumopathie interstitielle non spécifique.
Item 213 Anémie
C'est une cause d'anémie hémolytique auto-immune.
Item 239 Acrosyndromes
C'est une cause de phénomène de Raynaud secondaire.
Item 224 Hypertension artérielle de l'adulte et de l'enfant
C'est une cause d'HTA maligne via la crise rénale sclérodermique.
Item 273 Dysphagie
C'est une cause dysphagie non lésionnelle par troubles moteurs secondaires.
Item 283 Constipation
C'est une cause de constipation secondaire métabolique.
Item 305 Tumeurs de l'œsophage
C'est un facteur de risque de cancer épidermoïde de l'œsophage.
Références
Amylose AL
- En (très) bref
- Items où elle est abordée
- Item 90 Pathologie des glandes salivaires
- Item 95 Radiculalgie et syndrome canalaire
- Item 96 Neuropathies périphériques
- Item 212 Anémie
- Item 216 Syndrome hémorragique
- Item 234 Insuffisance cardiaque de l'adulte
- Item 235 Péricardite aiguë
- Item 261 Néphropathie glomérulaire
- Item 264 Insuffisance rénale chronique chez l’adulte et l’enfant
- Item 267 Troubles de l’équilibre acido-basique et désordres hydro-électrolytiques
- Item 275 Splénomégalie
- Item 283 Constipation
- Item 320 Myélome multiple des os
- Item 342 Malaise, perte de connaissance, crise comitiale chez l’adulte
- Item 347 Rétention aiguë d'urines
- Références
En (très) bref
Épidémiologie
L'incidence de l'amylose AL est autour de 1 pour 100 000 habitants. La prévalence est de 0.3 pour 100 000 habitants en France. L'âge moyen de survenue est autour de 60 ans. Les hommes sont plus fréquemment atteints avec un sex ratio de 1.5 hommes pour 1 femme.
Signes
L'amylose AL peut théoriquement atteindre tous les organes sauf le cerveau et le corps vitré. Les atteintes les plus fréquentes sont cardiaques et rénales.
Signes généraux
On peut voir une asthénie et une perte de poids.
Signes cardiaques
À terme, les dépôts s'accumulent et apparaît une cardiomyopathie restrictive. Elle peut être responsable d'une insuffisance cardiaque systolique ou diastolique, et de troubles de la conduction.
Signes rénaux
Les dépôts se concentrent dans le glomérule et donnent un syndrome néphrotique. Toutes les complications de ce dernier peuvent alors apparaître.
Signes neurologiques
20% des patient·e·s ont une polyneuropathie axonale longueur-dépendante qui s'accompagne donc de ses signes moteurs, sensitifs, et autonomes.
Signes hépatiques
On peut voir une hépatomégalie avec cholestase biologique anictérique. À noter qu'il n'y a pas d'insuffisance hépato-cellulaire associée.
Signes digestifs
L'atteinte digestive histologique est très fréquente mais les symptômes beaucoup moins. Au niveau du tube digestif bas, il peut y avoir des troubles du transit. Au niveau de la sphère buccale, l'infiltration peut être responsable d'un syndrome sec et d'une macroglossie.
Causes
Les signes de la maladie sont en lien avec des dépôts extra-cellulaires de chaînes légères d'une immunoglobuline monoclonale. Elles sont synthétisées par une prolifération monoclonale de plasmocytes.
Diagnostic/Examens complémentaires
Le diagnostic repose sur la biopsie, principalement des glandes salivaires accessoires. Elle permet la mise en évidence de dépôts amyloïdes à la coloration rouge Congo et de chaînes légères à l'immuno-histochimie.
Le bilan initial cherche notamment à caractériser les atteintes cardiaques, rénales, hépatiques et neurologiques.
Traitement
Les traitements actuels ne permettent pas de faire disparaître les dépôts amyloïdes déjà présents mais cherchent plutôt à diminuer leur prolifération. Il s'agit donc de chimiothérapies et thérapies ciblées visant les cellules B.
Iconographie
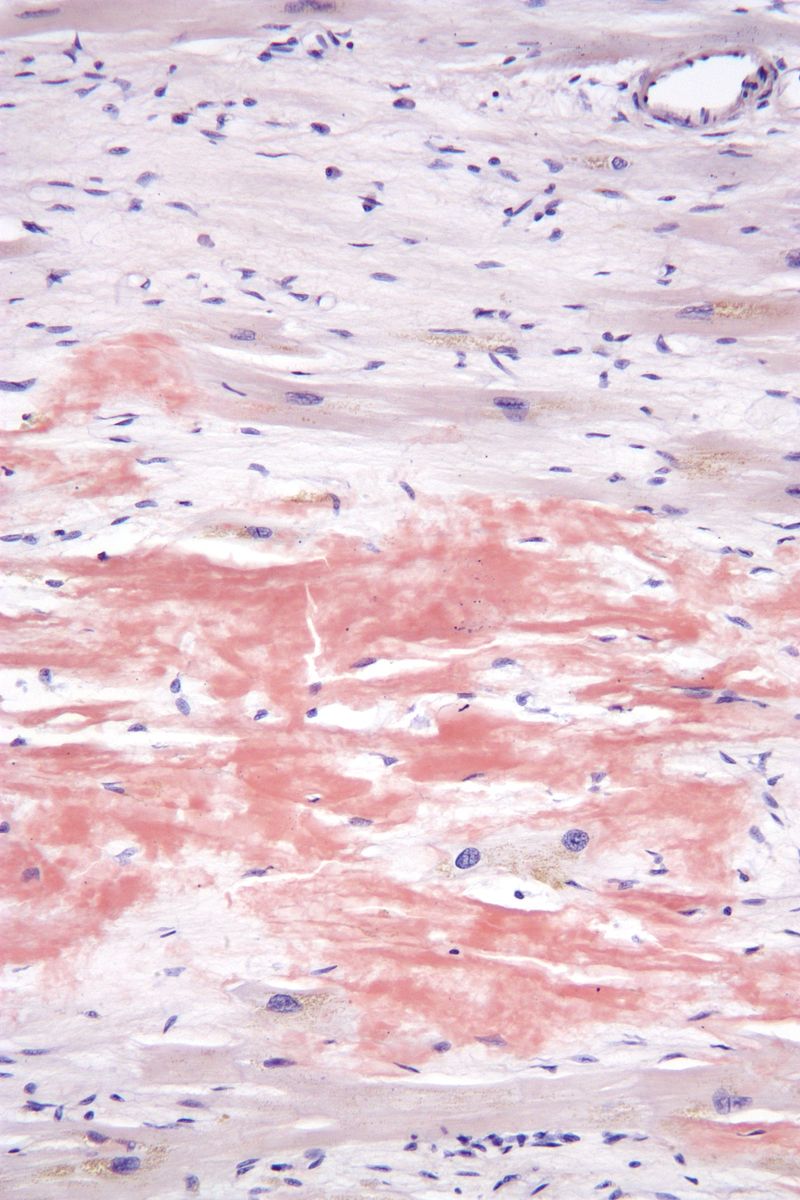
Items où elle est abordée
Item 90 Pathologie des glandes salivaires
C'est une cause de sialose.
Item 95 Radiculalgie et syndrome canalaire
C'est une cause de syndrome canalaire par augmentation du volume du contenu.
Item 96 Neuropathies périphériques
C'est une cause de polyneuropathie axonale longueur-dépendante.
Item 212 Anémie
Elle peut être responsable d'un asplénisme et donc d'une thrombocytose.
Item 216 Syndrome hémorragique
C'est une cause d'élévation du TCA et de baisse du TP par déficit en facteur X.
Item 234 Insuffisance cardiaque de l'adulte
C'est une cause d'insuffisance cardiaque par cardiomyopathie restrictive.
Item 235 Péricardite aiguë
C'est une cause de péricardite.
Item 261 Néphropathie glomérulaire
C'est une cause de syndrome néphrotique.
Item 264 Insuffisance rénale chronique chez l’adulte et l’enfant
C'est une cause d'insuffisance rénale chronique à reins non petits.
Item 267 Troubles de l’équilibre acido-basique et désordres hydro-électrolytiques
L'infiltration de la glande parathyroïde peut donner une hypocalcémie.
Item 275 Splénomégalie
C'est une cause de splénomégalie par fonction macrophagique.
Item 283 Constipation
C'est une cause constipation secondaire.
Item 320 Myélome multiple des os
Elle peut être associée au myélome multiple symptomatique.
Item 342 Malaise, perte de connaissance, crise comitiale chez l’adulte
C'est une cause dysautonomie
Item 347 Rétention aiguë d'urines
Elle peut être responsable d'un hypocontractilité vésicale myogène par infiltration.
Références
Amylose AA
En (très) bref
Épidémiologie
Grâce au progrès dans le diagnostic et le traitement des maladies inflammatoires, l'amylose AA se fait de plus en plus rare. L'incidence était de 0.1 pour 100 000 habitants au Royaume-Uni en 20081.
Signes
Les manifestations de la maladie peuvent se rapprocher de celles de l'amylose AL. L'atteinte la plus fréquente de l'amylose AA est l'atteinte rénale qui se traduit par un syndrome néphrotique.
Causes
La maladie est en lien avec des dépôts d'une protéine de l'inflammation, la protéine sérum amyloïde A.
C'est une complication des syndromes inflammatoires chroniques. On la rencontre donc dans des maladies inflammatoires chroniques :
- rhumatismales, comme la spondylarthrite ankylsante ou la polyarthrite rhumatoïde ;
- digestives, avec les MICI ;
- des infections chroniques, par exemple la tuberculose et la dilatation des bronches.
Diagnostic/Examens complémentaires
Le diagnostic repose sur la biopsie, principalement des glandes salivaires accessoires. Elle permet la mise en évidence de dépôts amyloïdes à la coloration rouge Congo et de protéine sérum amyloïde A à l'immuno-histochimie.
Traitement
Le traitement est celui du syndrome inflammatoire sous-jacent. Il permet de freiner l'évolution de la maladie et parfois de faire régresser les dépôts.
Iconographie
Items où elle est abordée
Item 185 Réaction inflammatoire
C'est une complication des syndromes inflammatoires prolongés.
Item 197 Spondyloarthrite
C'est une complication des spondyloarthrites.
Item 261 Néphropathies glomérulaires
C'est une cause de syndrome néphrotique.
Références
Connectivite mixte
En (très) bref
Épidémiologie
La prévalence de la connectivite mixte serait de 1 pour 100 000 habitants. Elle début le plus souvent entre 20 et 50 ans mais le diagnostic est souvent difficile. Elle atteint préférentiellement les femmes avec un sex ratio de 8 femmes pour 1 homme.
Signes
Un·e patient·e atteint·e de connectivite indifférenciée présente plusieurs signes rentrant dans les critères de classification de maladie auto-immune bien définie (lupus erythémateux disséminé, sclérodermie systémique, myosite) sans pour autant en remplir suffisamment pour être classé·e dans une de ces cases.
La connectivite mixte est un cas particulier de connectivite indifférenciée caractérisé par la présence d'anticorps anti-RNP.
La maladie associe des signes de plusieurs autres maladies auto-immunes systémiques au programme (comme le lupus) ou traité dans d'autres chapitres (commme la sclérodermie systémique).
Le syndrome de Sharp est une connectivite mixte (avec des anticorps anti-RNP donc) où la symptomatologie se limite à :
- un phénomène de Raynaud ;
- des doigts boudinés ;
- des arthralgies.
Causes
Les causes de la maladie sont inconnues. Comme pour beaucoup de maladies auto-inflammatoires, il y a très certainement un terrain génétique prédisposant non suffisant et des facteurs environnementaux.
Diagnostic/Examens complémentaires
UpToDate propose les critères de classification suivants qui font la synthèse de 4 propositions :
- des anticorps anti-RNP ;
- au moins 3 manifestations parmi :
- des doigts boudinés ;
- un phénomène de Raynaud ;
- une synovite ;
- une myosite ;
- une sclérose cutanée des doigts.
La difficulté du diagnostic consiste à distinguer la connectivite mixte d'une forme débutante d'une autre maladie auto-immune systémique.
Traitement
Le traitement est celui des manifestations de la maladie. Par exemple, des règles hygiéno-diététiques pour le phénomène de Raynaud, ou de l'hydroxychloroquine pour les arthralgies.
Items où elle est abordée
Item 192 Pathologies auto-immunes : aspects épidémiologiques, diagnostiques et principes de traitement
C'est une maladie auto-immune non spécifique d'organes.
Item 239 Acrosyndromes
C'est une cause de phénomène de Raynaud secondaire.
Références
Syndrome de chevauchement
En (très) bref
Épidémiologie
Nous n'avons pas facilement trouvé de données épidémiologique sur les différents syndromes de chevauchement.
Signes
Les patient·e·s atteint·e·s d'un syndrome de chevauchement remplissent les critères de classification de plusieurs maladies auto-immunes. Ce cas est à opposer aux connectivites mixtes où les patient·e·s ne présentent pas suffisamment de critère pour rentrer dans la case d'une maladie auto-immune. À noter qu'un lupus induit par un médicament donné pour une autre maladie auto-immune ne permet pas de caractériser un syndrome de chevauchement.
Certaines associations sont plus fréquentes que d'autres : lupus et polyarthrite rhumatoïde, sclérodermie et dermatomyosite.
Causes
Les causes de la maladie sont inconnues. Comme pour beaucoup de maladies auto-inflammatoires, il y a très certainement un terrain génétique prédisposant non suffisant et des facteurs environnementaux.
Diagnostic/Examens complémentaires
Ils recoupent entièrement ceux des maladies associés dans le syndrome de chevauchement.
Traitement
Ils recoupent ceux des maladies associés dans le syndrome de chevauchement.
Items où il est abordé
Item 192 Pathologies auto-immunes : aspects épidémiologiques, diagnostiques et principes de traitement
C'est l'association de plusieurs maladies auto-immunes
Références
Maladie de Behçet
- En (très) bref
- Items où elle est abordée
- Item 83 Infections et inflammations oculaires
- Item 91 Déficit neurologique récente
- Item 104 Sclérose en plaque
- Item 151 Méningites, méningoencéphalites, abcès cérébral chez l’adulte et l’enfant
- Item 193 Vascularites
- Item 225 Artériopathie de l’aorte, des artères viscérales et des membres inférieurs ; anévrysmes
- Item 340 Accidents vasculaires cérébraux
- Références
En (très) bref
Épidémiologie
La maladie de Behçet a une prévalence inférieure à 5 pour 100 000 habitants en France mais il existe des variations en fonction des pays : les régions les plus touchées sont l'Asie, l'Afrique du Nord et le Moyen-Orient. Elle survient le plus souvent autour de 30 ans. Elle touche autant les hommes que les femems avec un sex-ratio à 1.
Signes
C'est une maladie qui évolue par poussées qui peuvent associer :
- une aphtose dite bipolaire (intéressant bouche et sphère génitale) ;
- des arthralgies inflammatoires des grosses articulations ;
- des uvéites quasi systématiquement au moins postérieures et parfois antérieures également, non granulomateuses ;
- des méningites ou méningo-encéphalites ;
- des thromboses artérielles ou veineuses (mais pas d'embolie pulmonaire).
Causes
À l'inverse des autres vascularites du programme, la maladie de Behçet n'a pas de taille de vaisseaux de prédilection, elle peut tous les toucher.
Comme pour beaucoup de maladies auto-inflammatoires, la génétique joue un rôle. L'allèle HLA B*51 joue un rôle.
Diagnostic/Examens complémentaires
Les critères de classification permettent de retenir la maladie pour un score supérieur ou égal à 41.
| Item | Points |
|---|---|
| Aphtes buccaux | 2 |
| Aphtes génitaux | 2 |
| Atteinte oculaire | 2 |
| Atteinte cutanée | 1 |
| Atteinte vasculaire | 1 |
| Atteinte neurologique | 1 |
| Pathergy test positif | 1 |
Le pathergy test recherche l'apparition de lésions ulcérées caractéristiques à la piqûre.
Traitement
La colchicine est utile pour les atteintes cutanées et articulaires.
Les formes sévères de la maladie nécessitent un traitement immunosuppresseur, des corticoïdes en première intention.
Items où elle est abordée
Item 83 Infections et inflammations oculaires
C'est une cause d'uvéite antérieure aiguë avec hypopion mais aussi d'uvéite postérieure. C'est aussi une cause de sclérite.
Item 91 Déficit neurologique récente
C'est une cause d'atteinte du système nerveux central inflammatoire.
Item 104 Sclérose en plaque
C'est un diagnostic différentiel de névrite optique rétro-bulbaire.
Item 151 Méningites, méningoencéphalites, abcès cérébral chez l’adulte et l’enfant
C'est une cause de méningite aseptique inflammatoire.
Item 193 Vascularites
C'est une vascularite des vaisseaux de tout calibre.
Item 225 Artériopathie de l’aorte, des artères viscérales et des membres inférieurs ; anévrysmes
C'est une cause d'anévrisme de l'aorte abdominale.
Item 340 Accidents vasculaires cérébraux
C'est une cause de thrombose veineuse cérébrale.
Références
Maladie associée aux IgG4
En (très) bref
Épidémiologie
La maladie associée aux IgG4 a une prévalence faible de l'ordre 0.1 pour 100 000 habitants en France. Elle survient le plus souvent autour de 55 ans. Elle touche préférentiellement les hommes.
Signes
Les manifestations de la maladie sont en lien avec l'envahissement des tissus par des cellules du système immunitaires jusqu'à former d'authentiques pseudo-tumeurs. Ces organomégalies peuvent être responsables de compression des organes de voisinage. À terme, les tissus peuvent se fibroser.
Les signes les plus fréquents ou les plus en lien avec le programme sont donc :
- des douleurs abdominales en lien avec une pancréatite aiguë auto-immune ;
- un ictère en lien avec une cholangite sclérosante secondaire ;
- des gonflements des glandes salivaires et lacrymales ;
- des adénomégalies, plutôt profondes ;
- une fibrose rétro-péritonéale ;
- une néphropathie interstitielle chronique.
Causes
Les causes de la maladie sont inconnues. Comme pour beaucoup de maladies auto-inflammatoires, il y a très certainement un terrain génétique prédisposant non suffisant et des facteurs environnementaux. À noter qu'aucune forme familiale n'a été mise en évidence à ce jour.
Parmi les cellules inflammatoires infiltrant les tissus, on trouve des plasmocytes qui sécrètent principalement des IgG4, donnant le nom à la maladie.
Diagnostic/Examens complémentaires
Le diagnostic repose sur l'association de plusieurs critères :
- la mise en évidence, clinique ou radiologique, d'une organomégalie :
- des anomalies anatomo-pathologiques caractéristiques ;
- une élévation du taux d'IgG4 dans le sang ;
- l'élimination des diagnostics différentiels.
Des critères de classification ont été élaborées par l'American College of Rheumatology et l'European League Against Rheumatism en 20191. Ils sont basés sur :
- la mise en évidence, clinique, radiologiquee ou anatomo-pathologique, d'une atteinte évocatrice ;
- un certain nombre de critères d'exclusion, plus évocateurs d'un diagnostic différentiel ;
- un système de points combinant des critères cliniques, radiologiques et anatomo-pathologiques.
Traitement
Le traitement repose sur la corticothérapie. Un geste chirurgical est parfois utile en cas de compression par les pseudo-tumeurs.
Items où elle est abordée
Item 90 Pathologies des glandes salivaires
C'est une cause de sialose.
Item 218 Éosinophilie
C'est une cause systémique d'éosinophilie.
Item 262 Néphropathies interstitielles chroniques
C'est une cause de néphropathie intersitielle chronique dysimmunitaire.
Item 278 Ictère
C'est une cause de cholangite sclérosante secondaire.
Item 258 Pancréatite aiguë
C'est une cause de pancréatite aiguë.
Références
Syndrome cave supérieur
En (très) bref
Épidémiologie
L'épidémiologie du syndrome cave supérieur épouse celle de ses causes.
Signes
Le syndrome cave supérieur traduit la compression de la veine cave supérieure. Les signes sont en lien avec la stase veineuse en amont :
- œdème en pèlerine touchant les bras, la face, le cou, et le haut du thorax. On peut alors voir un comblement des creux sus-claviculaires ;
- turgescence jugulaire ;
- cyanose du territoire drainé par la veine cave supérieure.
Les signes d'alerte sont les 3 D :
- dyspnée ;
- dysphagie ;
- dysphonie.
Causes
Toutes les causes de compression de la veine cave supérieure peuvent en être à l'origine.
On retiendra surtout les causes tumorales qu'ils s'agissent d'un envahissement local d'une tumeur solide ou des adénopathies d'une hémopathie.
Les matériels endo-vasculaires peuvent provoquer des thromboses qui peuvent être à l'origine d'un authentique syndrome cave supérieur.
Diagnostic/Examens complémentaires
Le bilan ne doit pas retarder la prise en charge et peut comporter :
- un angioscanner cervico-thoracique où l'injection doit être faite via les 2 bras ;
- NFS, bilan de coagulation, bilan rénal.
Traitement
Il associe :
- une mise en position demie-assies ;
- une oxygénothérapie ;
- une anticoagulation préventive ;
- le traitement de la cause qui peut commencer par une corticothérapie.
Iconographie
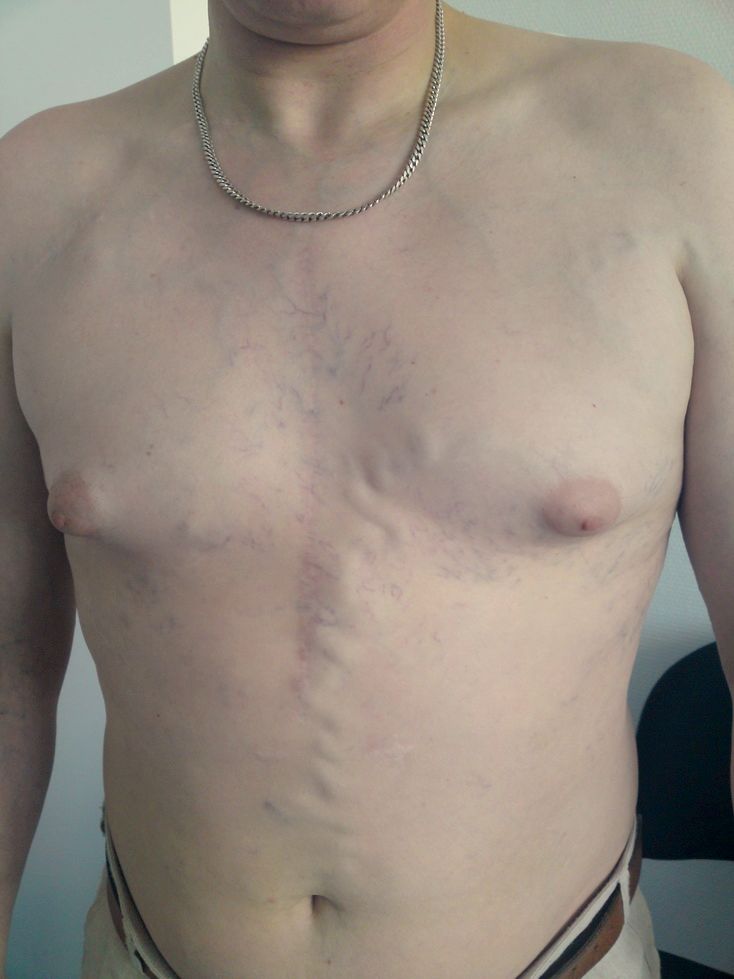
Items ou il est abordé
Item 309 Tumeurs du poumon
C'est une signe d'invasion loco-régionale des cancers du poumons, en particulier des cancers à petites cellules.
Item 319 Lymphomes malins
C'est une urgence compressive dans les lymphomes qui touchent les ganglions médiastinaux.
Item 338 Anaphylaxie
C'est un diagnostic différentiel de l'anaphylaxie.
Références
Syndrome de lyse tumorale
- En (très) bref
- Items où il est abordé
- Item 258 Élévation de la créatininémie
- Item 267 Troubles de l’équilibre acido-basique et désordres hydro-électrolytiques
- Item 292 Traitement des cancers : principales modalités, classes thérapeutiques et leurs complications majeures. La décision thérapeutique pluridisciplinaire et l’information du malade
- Item 315 Leucémies aiguës
- Item 319 Lymphomes malins
- Références
En (très) bref
Épidémiologie
Nous n'avons pas facilement trouvé de données épidémiologique sur les différents syndromes de chevauchement.
Signes
Le syndrome de lyse tumorale traduit la destruction de cellules tumorales et le relargage de leur contenu dans la circulation sanguine.
Les signes biologiques suivent la même logique que dans la rhabdomylose :
- une hyperkaliémie, une hyperphosphatémie et une hyperuricémie, en lien avec le relargage du contenu des myocytes ;
- une hypocalcémie consécutive à une chélation par les ions phosphates ;
- une acidose métabolique à trou anionique augmentée ;
- une insuffisance rénale aiguë en lien avec la myoglobinurie et la précipitation du phosphate et de l'acide urique.
Les signes cliniques suivent de ces perturbations :
- des troubles du rythme et de la conduction cardiaque ;
- des troubles neurologiques en lien avec l'hypocalcémie.
Causes
Les tumeurs les plus promptes à ce syndrome sont celles à division rapide :
- les hémopathies, et notamment les lymphomes non hodgikiniens de haut grade (dont le lymphome de Burkitt), les leucémies aiguës ;
- les cancers solides, et principalement les cancers du poumon à petites cellules, les tumeurs germinales, les neuroblastomes.
Des facteurs de risque tels qu'une insuffisance rénale augmentent la probabilité de faire un syndrome de lyse tumorale clinique.
La lyse peut être spontanée quand le temps de doublement de la masse tumorale est faible. Elle peut aussi être déclenchée par des traitements spécifiques du cancer, notamment les chimiothérapies qui ciblent des étapes du cycle de division cellulaire.
Diagnostic/Examens complémentaires
Des critères diagnostiques ont été proposés.
| Item | Points |
|---|---|
| Calcémie < 1.75 mmol/L ou baisse > 25% | 1 |
| Potassium > 6 mmol/L ou hausse > 25% | 1 |
| Phosphates > 1.45 mmol/L ou hausse > 25% | 1 |
| Acide urique > 476 µmol/L ou hausse > 25% | 1 |
2 points suffisent à retenir un syndrome de lyse tumorale biologique.
On parle de syndrome de lyse tumorale clinique quand un des signes suivants s'ajoute au syndrome de lyse tumorale biologique :
- une élévation de la créatinine ;
- des convulsions ;
- une arythmie cardiaque.
En fonction de l'intensité de ces 3 manifestations, on peut grader le syndrome de lyse tumorale clinique.
Traitement
Le traitement préventif inclue :
- l'hydratation intra-veineuse pour maintenir une bonne diurèse ;
- le traitement de certains troubles hydro-électrolytiques : l'hyperuricémie et l'hyperkaliémie ;
- le respect d'autres troubles hydro-électrolytiques : l'hypocalcémie (dont la correction risquerait de majorer la précipitation du phosphates), le non-recours à l'alcalinisation des urines dont le rôle est controversé ;
- et bien sûr le traitement spécifique du cancer.
Le traitement curatif ajoutera aux mesures préventives la discussion des options suivantes :
- l'expuration extra-rénale ;
- des diurétiques pour maintenir une bonne diurèse ;
- des chélateurs du phosphate.
Items où il est abordé
Item 258 Élévation de la créatininémie
C'est une cause d'insuffisance rénale aiguë organique.
Item 267 Troubles de l’équilibre acido-basique et désordres hydro-électrolytiques
C'est une cause d'hyperkaliémie.
Item 292 Traitement des cancers : principales modalités, classes thérapeutiques et leurs complications majeures. La décision thérapeutique pluridisciplinaire et l’information du malade
Les anthracyclines, l'étoposide et le méthotrexate sont particulièrement à risque de syndrome de lyse tumorale.
Item 315 Leucémies aiguës
C'est une complication des leucémies aiguës.
Item 319 Lymphomes malins
C'est une complication de certains lymphomes de haut grade.
Références
- Chapitre du référentiel de Médecine intensive, réanimation, urgences et défaillances viscérales aiguës édition 2018
- UpToDate
- UpToDate bis
Syndrome d'activation macrophagique
En (très) bref
Épidémiologie
Le syndrome d'activation macrophagique, aussi appelé lymphohistiocytose hématophagocytaire dans les pays anglo-saxons, est une maladie rare qui peut être primaire ou secondaire.
La forme primaire est une maladie rare dont l'incidence est mal connue mais probablement inférieure à 1 pour 100 000 habitants. C'est une maladie héréditaire qui concerne l'enfant avec un sex-ratio proche de un.
Les formes secondaires peuvent toucher des adultes. Cela reste une maladie très rare dont l'incidence est mal caractérisée.
Signes
Les différents signes apparaissent généralement brutalement.
À la clinique, on retrouvera :
- une fièvre dans 100% des cas ;
- une altération de l'état général ;
- une hépatosplénomégalie ;
- des adénopathies.
Les signes biologiques sont plus riches :
- des cytopénies dans 100% des cas (la leucopénie est moins fréquente que l'anémie normocytaire arégénérative et la thrombopénie) ;
- une hyperferritinémie ;
- une hypertriglycéridémie sans hypercholestérolémie ;
- une cytolyse hépatique ;
- une baisse du fibrinogène.
Causes
Les formes primaires sont liées à des maladies génétiques rarissimes qui vont bien au-delà des buts de cet ouvrage.
Les formes secondaires peuvent être rencontrées dans :
- des néoplasies, et plus particulièrement les hémopathies avec en tête des lymphomes de haut grade ;
- des maladies auto-inflammatoires, comme le lupus érythémateux disséminé, les arthrites juvéniles, ou le DRESS ;
- des infections, toutes peuvent être en cause, virus en tête.
Les mécanismes cellulaires en cause ne sont pas encore entièrement élucidés. L'activation macrophagique semble être secondaire à un autre primum movens qui sera une anomalie des lymphocytes T ou NK. L'hypertriglycéridémie est liée à l'inihibition de la lipoprotéine lipase par des cytokines inflammatoires.
Diagnostic/Examens complémentaires
L'examen clé est le myélogramme. Il montrera une hémophagocytose qui n'est pas pour autant pathognomonique de la maladie, ni 100% sensible.
Traitement
Le traitement est :
- symptomatique, en corrigeant les cytopénies ;
- étiologique, en agissant sur la cause sous-jacente ;
- spécifique, avec parfois un recours aux immunosuppresseurs.
Items où il est abordé
Item 115 Toxidermies
Le DRESS peut s'accompagner d'un syndrome d'activation macrophagique.
Item 213 Anémie
Le syndrome d'activation macrophagique est une cause d'anémie normocytaire arégénérative à moelle riche.
Item 214 Thrombopénie
Le syndrome d'activation macrophagique est une cause de thrombopénie.
Item 275 Splénomégalie
Le syndrome d'activation macrophagique est une cause de splénomégalie.
Item 296 Agranulocytose médicamenteuse
Le syndrome d'activation macrophagique est une cause de neutropénie.
Item 319 Lymphomes malins
Les lymphomes de haut grade peuvent s'accompagner d'un syndrome d'activation macrophagique.
Références
- Karras, A., & Hermine, O. (2002). Syndrome d’activation macrophagique. La revue de médecine interne, 23(9), 768-778.
- Gonzalez, F., Vincent, F., & Cohen, Y. (2009). Syndrome d’activation macrophagique d’origine infectieuse: étiologies et prise en charge. Réanimation, 18(4), 284-290.
- Syndrome d’activation macrophagique. (2014). La Revue de Médecine Interne, 35, A24–A30. doi:10.1016/j.revmed.2014.03.009
Maladie de Gaucher
En (très) bref
Épidémiologie
La maladie de Gaucher est une maladie génétique à transmission autosomique dominante. La prévalence est d'environ 1 pour 100 000 habitants. La maladie est plus fréquente dans certaines sous-populations comme les juifs ashkénazes. L'âge médian au diagnostic est autour de 20 ans.
Signes
Il existe 3 formes de la maladie. Seul le type 1, le plus fréquent (90% des cas) et le plus en lien avec le programme du 2e cycle, sera évoqué ici.
La maladie de Gaucher fait partie de la famille des maladies de surcharge. Les signes sont en lien avec l'accumulation d'un lipide dans les lyzosomes. On observe donc :
- une organomégalie :
- une splénomégalie qui peut être majeure ;
- une hépatomégalie sans cirrhose ;
- des cytopénies : surtout anémie et thrombopénie ;
- des signes osseux :
- des douleurs prédominant aux membres inférieurs et au bassin
- des fractures
- des ostéonécroses aseptiques
Tout cela résulte en une asthénie. Chez l'enfant, on observe un retard pubertaire.
Cela étant dit, l'expression de la maladie est très variable, jusqu'à des formes quasi asymptomatiques.
Causes
La maladie de Gaucher est lié à un déficit en une enzyme des lyzosomes, la bêta-glucocérébrosidase. Cette enzyme catalyse la dégradation d'un lipide, le glucocérébroside, qui est une constituant des membranes plasmiques. Les signes sont en lien avec l'accumulation de macrophages chargés de ce lipide dans différents tissus et l'inflammation résultante.
Diagnostic/Examens complémentaires
Le diagnostic repose sur la mise en évidence d'une activité réduite de la bêta-gluocérébrosidase. On analyse ensuite le gène de cette enzyme.
Le bilan initial vise à faire la cartographie des lésions de la maladie
Traitement
Il existe 2 stratégies de traitement spécifique :
- l'enzymothérapie substitutive qui consiste à administrer une enzyme permettant la dégradation du glucocérébroside ;
- la réduction de substrat qui consiste à diminuer la sythèse de glucocérébroside.
Les indications se discutent en RCP. Les autres traitements sont symptomatiques.
Items où elle est abordée
Item 275 Splénomégalie
La maladie de Gaucher est une cause splénomégalie.
Item 296 Agranulocytose médicamenteus
La maladie de Gaucher est une cause de neutropénie.
Références
Hypertension pulmonaire
- En (très) bref
- Items où elle est abordée
- Références
En (très) bref
Épidémiologie
L'hypertension pulmonaire du groupe 1, l'hypertension artérielle pulmonaire, a une prévalence autour de 2 pour 100 000. Les formes idiopathiques surviennent le plus souvent entre 30 et 50 ans.
Signes
Le principal symptôme est la dyspnée d'aggravation progressive.
À l'examen clinique, on peut trouver des signes direct d'hypertension pulmonaire :
- un éclat du B2 ;
- un souffle d'insuffisance tricuspide, holosystolique, majoré à l'inspiration profonde.
Dans les formes les plus évoluées de la maladie, apparaissent de signes d'insuffisance cardiaque droite :
- tachycardie et galop ;
- reflux hépato-jugulaire puis turgescence jugulaire ;
- hépatomégalie ;
- œdème des membres inférieurs.
Causes
Les hypertensions pulmonaires sont classées en 5 catégories, homogènes pour leur présentation clinico-biologique et leur traitement.
Groupe 1 Hypertension artérielle pulmonaire
Des modifications vasculaires élèvent les résistance artérielles pulmonaires au-delà de 2 unités de Wood. C'est à ce groupe qu'est réservée l'acronyme d'HTAP pour hypertension artérielle pulmonaire. On y trouve 3 entités :
- les HTAP idiopathiques, sans antécédents familiaux ;
- les HTAP héritables, avec antécédents familiaux ;
- les HTAP associées à des médicaments (dont le benfluorex) ou d'autres maladies (dont la sclérodermie).
Groupe 2 Hypertension pulmonaire des cardiopathies gauches
L'augmentation des pressions dans les cavités gauches finit par retentir sur le poumon et à terme les cavités droites. L'insuffisance cardiaque gauche est la première cause d'hypertension pulmonaire.
Groupe 3 Hypertension pulmonaire des insuffisances respiratoires
L'hypoxie tissulaire chronique et l'altération du parenchyme pulmonaire peuvent à terme donner de l'hypertension pulmonaire.
Groupe 4 Hypertension pulmonaire post-embolique
On peut les considérer comme un syndrome post-thrombotique pulmonaire.
Groupe 5 Hypertension pulmonaire multifactorielle ou indéterminée
C'est une catégorie pour rassembler les hypertensions pulmonaires qui ne rentrent pas dans un des 4 groupes précédents.
À ces 5 catégories, se superposent une 2e classification basé sur cathétérisme cardiaque et la pression artérielle pulmonaire occluse (PAPO). Cette mesure permet d'approximer la pression régnant dans l'oreillette gauche. On distingue donc :
- les hypertensions artérielles post-capillaires où la PAPO est élevée : c'est l'augmentation des pressions dans les cavités gauches qui, mécaniquement, augmente la pression artérielle pulmonaire (groupe 2) ;
- les hypertensions artérielles pré-capillaires où la PAPO n'est pas élevée et où les résistances vasculaires pulmonaires sont supérieures à 2 unités de Wood : c'est une maladie du poumon en lui-même (groupe 1, 3, 4 et 5).
Diagnostic/Examens complémentaires
Une hypertension pulmonaire est définie par une pression artérielle pulmonaire moyenne supérieure à 22 mmHg1.
L'examen de première intention est donc l'échographie trans-thoracique qui sera complétée par un cathétérisme cardiaque pour obtenir un diagnostic de certitude.
Pour le bilan étiologique, certains examens systématiques compléteront l'interrogatoire et l'examen clinique :
- une scintigraphie pulmonaire de ventilation/perfusion pour éliminer une cause post-embolique ;
- un scanner thoracique qui pourra être injecté ;
- des épreuves fonctionnelles respiratoires ;
- des examens biologiques (bilan hépatique, sérologies virales, bilan auto-immun).
Traitement
Le traitement est complexe et peut faire appel à divers vasodilatateurs.
Items où elle est abordée
Item 208 Insuffisance respiratoire chronique
C'est une complication des insuffisances respiratoires chroniques.
Item 209 Bronchopneumopathie chronique obstructive chez l’adulte
L'oxygénothérapie longue durée peut limiter le risque d'hypertension pulmonaire chez les patient·e·s BPCO.
Item 226 Thrombose veineuse profonde et embolie pulmonaire
C'est une complication chronique des embolies pulmonaires.
Item 342 Malaise, perte de connaissance, crise comitiale chez l’adulte
C'est une cause de syncope cardiaque par obstacle mécanique.
Références
- Site de RespiFil
- Protocole National de Diagnostic et de Soins
- UpToDate
- Chapite du référentiel de pneumologie édition 20152
L'hypertension pulmonaire était un item du programme du deuxième cycle jusqu'en 2020.
Syndrome de Brugada
En (très) bref
Épidémiologie
La prévalence du syndrome de Brugada varie selon les pays : plus fréquent dans les pays asiatiques ainsi que leur diaspora. En France, elle s'établit entre 20 et 50 pour 100 000 habitants. Il se déclare à l'âge adulte et, s'il cause une mort subite, c'est autour de 40 ans. Il touche 4 hommes pour 1 femme.
Fun fact, ce syndrome est associé à la schizophrénie.
Signes
ECG
Il existe plusieurs types :
- Sus-décalage du segment ST en dôme en V1-V2 donnant un aspect bloc de branche droit au QRS. Ondes T souvent négatives dans ces dérivations ;
- Idem mais le sus-décalage est en forme de selle.
Les complications ECG incluent :
- tachycardie ventriculaire ;
- fibrillation ventriculaire ;
- bloc auriculo-ventriculaire complet ;
- dissociation électro-mécanique.
Clinique
Le syndrome de Brugada peut être responsable de lipothymies, syncopes et, à l'extrême, de mort subite.
Causes
C'est une maladie génétique à transmission autosomique dominante à pénétrance variable. Les gènes en cause codent pour des canaux sodiques. Cependant, la pénétrance incomplète indique la mise en jeu de mécanismes supplémentaires non complètement élucidés (anomalie d'autres canaux sodiques, altération de la microstructure du ventricule droit).
Il semble exister des facteurs déclenchants : fièvre, et substances (cocaïne notamment).
Diagnostic/Examens complémentaires
S'il n'y a pas de signes cliniques, on parle d'aspect Brugada. Le syndrome de Brugada peut donc se définir par l'association de signes ECG et de signes cliniques, en l'absence d'anomalie cardiaque structurelle. Le diagnostic peut aussi reposer sur l'utilisation de score.
Le diagnostic peut s'appuyer sur des tests de provocation en milieu hospitalier.
Traitement
Il n'existe pas de traitements spécifiques. La prise en charge est avant tout préventive et vise à l'éviction des facteurs déclenchants. À l'instar de la myasthénie, il existe un site internet répertoriant les médicaments à éviter.
Pour les patient·e·s les plus à risque, on peut proposer un défibrillateur implantable pour récupérer les fibrillations ventriculaires.
Iconographie
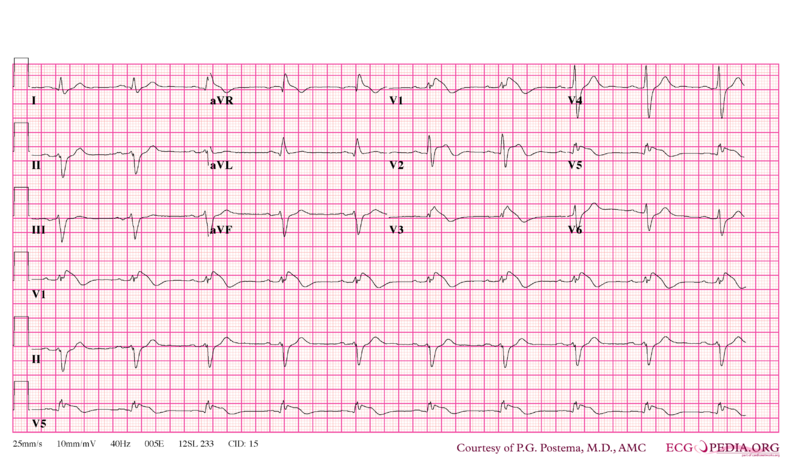
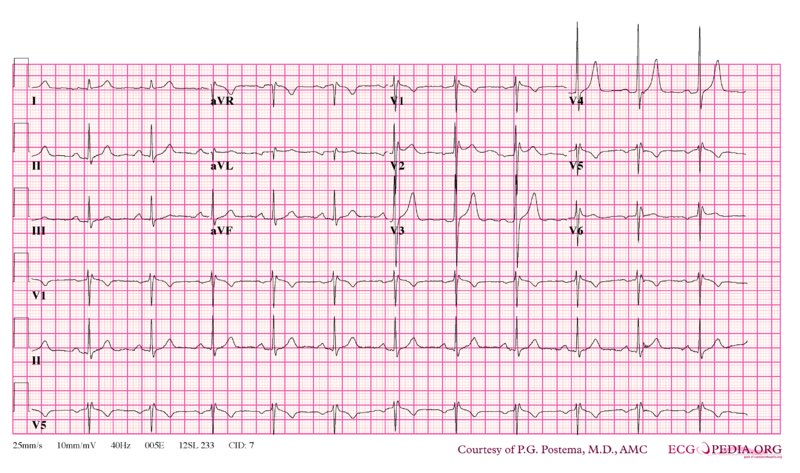
Items où il est abordé
Item 256 Aptitude au sport chez l’adulte et l’enfant ; besoins nutritionnels chez le sportif
C'est une contre-indication à la pratique sportive. À noter que ça n'en est pas une selon le PNDS.
Item 342 Malaise, perte de connaissance, crise comitiale chez l’adulte
Devant un malaise, c'est une anomalie ECG justifiant d'un avis cardiologique dans les 24 heures.
Références
Myocardite
En (très) bref
Épidémiologie
L’incidence des myocardites serait de 5 pour 100 000 habitants avec une légère prédominance masculine. Ces chiffres sont largement influencés par la diversité des causes sous-jacentes.
Signes
La myocardite a avant tout une définition histologique : l’inflammation du myocarde. La présentation clinique et son intensité vont dépendre de la cinétique d’installation et de la topographie de cette inflammation.
La présentation peut être aiguë et prendre différentes formes :
- un syndrome coronarien aigu à coronaires saines ;
- une insuffisance cardiaque aiguë ou rapidement progressive en quelques semaines, à l’extrême un choc cardiogénique ;
- des troubles du rythme pouvant conduire à une mort subite.
La présentation peut aussi être chronique et se rapprocher alors de l’insuffisance cardiaque chronique.
Causes
Les causes de myocardite sont extrêmement variées. Parmi les causes les plus fréquentes ou les plus en lien avec le programme on retrouve :
- les causes infectieuses. À peu près tous les germes peuvent être en cause, les virus en tête ;
- l’hyperéosinophilie ;
- la maladie coeliaque ;
- la dysplasie ventriculaire droite arythmogène ;
- la sarcoïdose ;
- les maladies auto-immunes dont le lupus érythémateux disséminé ;
- les toxiques dont la cocaïne.
Diagnostic/Examens complémentaires
Le diagnostic repose sur un faisceau d’arguments :
- des anomalies ECG ;
- une élévation des troponines ;
- des anomalies à l’imagerie où l’IRM a une place clé ;
- une biopsie dans les cas où le diagnostic est difficile.
Les examens complémentaires seront aussi guidés par la recherche de la cause.
Traitement
Le traitement comprend une part commune à toutes les myocardites (celui de l’insuffisance cardiaque et des troubles du rythme) et le traitement de la cause.
L’activité physique est à éviter à la phase aiguë, de même que les AINS.
Items où elle est abordée
Item 115 Toxidermies
La myocardite est une des manifestations viscérales du DRESS.
Item 164 Exanthèmes fébriles de l’enfant
La maladie de Kawasaki peut s’accompagner d’une myocardite.
Item 166 Grippe
La myocardite est une complication de la grippe.
Item 175 Voyage en pays tropical
La typhoïde peut s’accompagner de manifestations en lien avec des toxines dont la myocardite fait partie.
Item 194 Lupus systémique. Syndrome des anti-phospholipides
La myocardite est une des manifestations du lupus (mais la péricardite reste une atteinte cardiaque plus fréquente).
Item 235 Péricardite
Une péricardite peut s’accompagner et se compliquer d’une myocardite que l’on détectera via une augmentation des troponines.
Item 332 Etats de choc
La myocardite est une cause de choc cardiogénique.
Item 345 Malaise grave du nourrisson et mort inattendue du nourrisson
La myocardite est une cause de mort inattendue du nourrisson.
Références
Cholangite biliaire primitive
En (très) bref
Épidémiologie
La prévalence de la cholangite biliaire primitive est autour de 30 cas pour 100 000 habitants. Elle touche 9 femmes pour 1 homme. L'âge médian au diagnostic est de 50 ans.
Signes
Les signes de la maladie les plus fréquents sont l'asthénie et le prurit. On peut également observer :
- un syndrome sec ;
- des arthralgies ;
- une douleur de l'hypochondre droit.
Dans les formes évoluées, on peut voir :
- un ictère ;
- des xanthomes péri-orbitaires ;
- une hépatomégalie.
À terme et sans traitement, la maladie évolue vers une cirrhose. L'ancien nom de la maladie était d'ailleurs la cirrhose biliaire primitive.
La cholangite biliaire primitive peut s'associer à d'autres maladies auto-immunes : dysthyroïdie, maladie cœliaque, syndrome de Gougerot-Sjögren, ou sclérodermie.
Causes
Les causes de la maladie sont inconnues. Comme pour beaucoup de maladies auto-inflammatoires, il y a très certainement un terrain génétique prédisposant non suffisant et des facteurs environnementaux.
Diagnostic/Examens complémentaires
La diagnostic est biologique et repose sur l'association :
- d'une cholestase ;
- des auto-anticorps spécifiques : les anti-mitochondries de type 2 ou certains types d'anti-nucléaires.
Les autres signes biologiques observables sont :
- une élévation modérée des transaminases ;
- une hypercholestérolémie prédominant sur le HDL-cholestérol ;
- une augmentation polyclonale des IgM.
La cholangite biliaire primitive peut s'associer à une hépatite auto-immune dans le cadre d'un syndrome de chevauchement. Il faut y penser devant une élévation des transaminases supérieure à 5N ou une augmentation polyclonale des IgG.
Le bilan initial inclue également une échographie hépatique et une mesure élastométrique du foie.
À noter que la biopsie hépatique n'est généralement pas nécessaire.
Traitement
Le traitement de première ligne de référence est l'acide deoxyursocholique per os.
Items où elle est abordée
Item 278 Ictère
C'est une cause d'ictère à bilirubine conjuguée cholestatique par obstacle sur les petits canaux.
Item 279 Cirrhose
C'est une cause, rare, de cirrhose.
Item 304 Tumeurs du foie
C'est un facteur de risque de cholangiocarcinome.
Références
Cholangite sclérosante primitive
En (très) bref
Épidémiologie
La prévalence de la cholangite sclérosante primitive est d'environ 8 pour 100 000 habitants. Elle débute généralement autour de 40 ans et touche 2 hommes pour 1 femme.
Signes
La maladie peut prendre beaucoup de formes et doit donc être suspectée dans de nombreuses conditions une fois les diagnostics plus fréquents éliminés. Les signes associent inconstamment :
- des signes biliaires : ictère, prurit, angiocholite ;
- des signes en lien avec une hépatopathie chronique ;
- des signes biologiques : une cholestase, une augmentation de la bilirubine.
S'associe souvent une maladie inflammatoire chronique de l'intestin : 50% des patient·e·s atteint·e·s de cholangite sclérosante primitive ont une MICI, une rectocolite hémorragique dans 75% des cas.
En dehors de la cirrhose et toutes ses conséquences, une des complications de la maladie est le cholangiocarcinome.
Causes
Les causes de la maladie sont inconnues. Comme pour beaucoup de maladies auto-inflammatoires, il y a très certainement un terrain génétique prédisposant non suffisant et des facteurs environnementaux.
Diagnostic/Examens complémentaires
Le diagnostic demande d'éliminer les causes de cholangite sclérosante secondaire (qui incluent notamment la maladie associée aux IgG4).
L'examen clé du diagnostic est la cholangio-IRM.
Le diagnostic de gravité demande d'évaluer le niveau de cirrhose du foie.
Traitement
En dehors des traitements du prurit, le seul traitement à l'efficacité démontrée à ce jour est la transplantation hépatique, qui est réservée au cas les plus sévères.
Items où elle est abordée
Item 278 Ictère
C'est une cause d'ictère.
Item 282 Maladies inflammatoires chroniques de l'intestin chez l'adulte
Elle est fréquemment associée aux MICI.
Item 304 Tumeurs du foie, primitives et secondaires
Le cholangiocarcinome est une complication de la cholangite sclérosante primitive.
Références
Lichen plan
- En (très) bref
- Items où il est abordé
- Item 58 Sexualité normale et ses troubles
- Item 116 Prurit
- Item 155 Infections cutanéo-muqueuses et des phanères, bactériennes et mycosiques de l’adulte et de l’enfant
- Item 162 Infections sexuellement transmissibles (IST) : gonococcies, chlamydioses, syphilis, papillomavirus humain (HPV), trichomonose
- Item 168 Infections à herpès virus du sujet immunocompétent
- Item 298 Tumeurs de la cavité buccale, naso-sinusiennes et du cavum, et des voies aérodigestives supérieures
- Item 302 Tumeurs cutanées, épithéliales et mélaniques
- Références
En (très) bref
Épidémiologie
La prévalence de la maladie serait d'environ 1000 pour 100 000 habitants. L'âge moyen de survenue est entre 30 et 60 ans. Il touche préferentiellement les femmes avec un sex ratio de 1.5 femmes pour 1 homme.
Signes
Le lichen plan peut atteindre la peau et les muqueuses.
Signes cutanés
La lésion typique est une papule violacée, polygonale à sommet plat. Elle est prurigineuse. Les lésions individuelles mesurent 2 à 4 mm mais peuvent s'agglomérer.
Les localisations les plus fréquentes sont les chevilles et les faces de flexion des poignets.
À noter qu'il peut exister, comme dans le psoriasis, un phénomène de Koebner où des petits traumatismes induisent des lésions.
Ils existent de nombreux variants du lichen plan (hypertrophique, annulaire, bulleux, actinique, etc.) qui dépassent le cadre de ce chapitre.
Signes muqueux
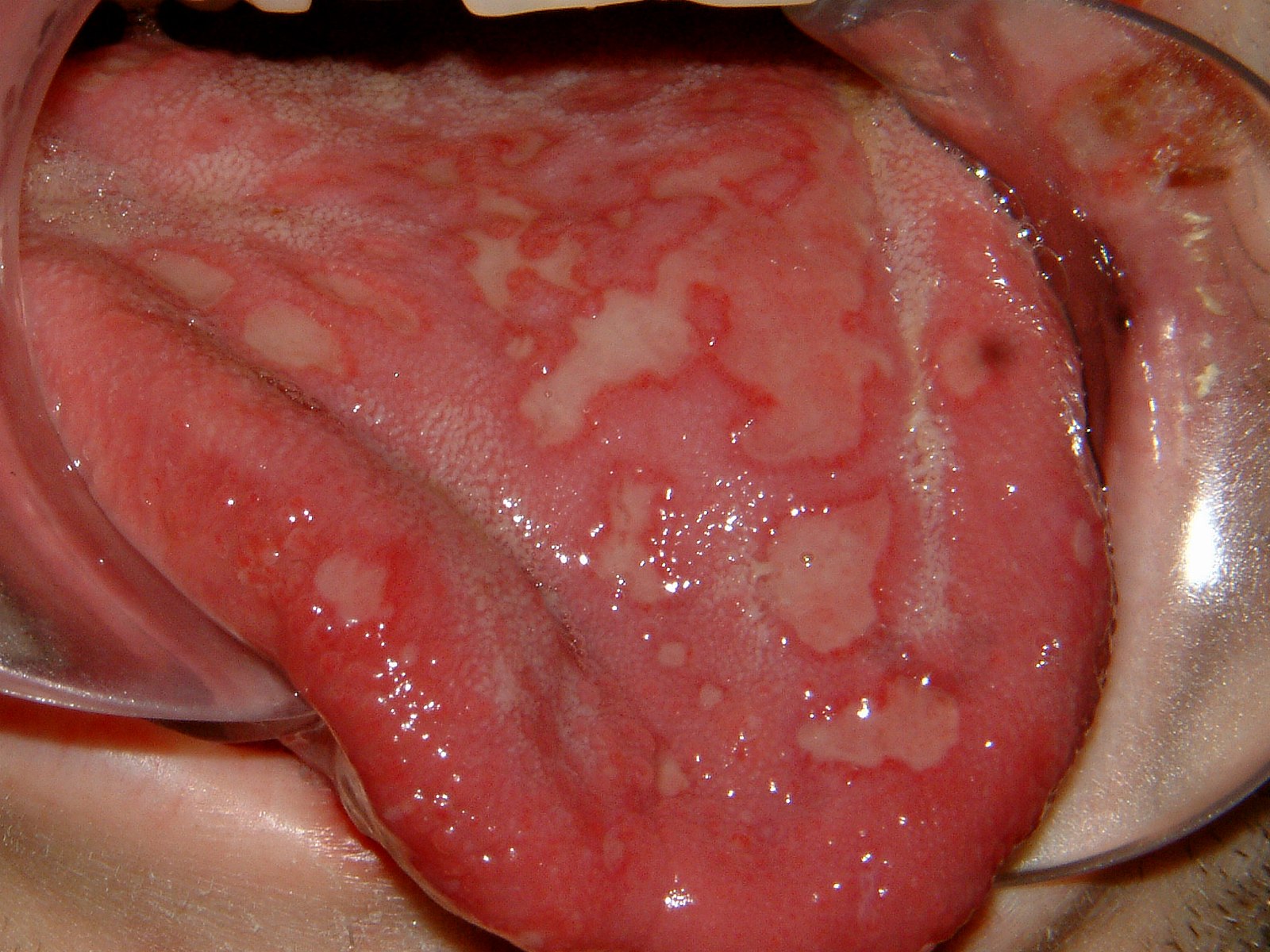
Au niveau de la muqueuse buccale, la lésion typique est la strie de Wickham, une lésion blanche linéaire et réticulé. D'autres lésions, notamment érosives, peuvent être rencontrées.
Au niveau des muqueuses génitales, on peut rencontré des lésions ressemblant aux papules violacées cutanées.
La muqueuse œsophgienne peut aussi être atteinte, responsable d'un tableau de dysphagie ou d'odynophagie.
À noter qu'il existe des syndromes de chevauchement avec la pemphigoïde et le lupus érythémateux disséminé.
Causes
La cause précise n'est pas connue. On sait que c'est une maladie médiée par l'immunité avec un rôle appuyé des cellules T CD8+. Certains médicaments peuvent être inducteurs.
Diagnostic/Examens complémentaires
Du fait des diagnostics différentiels qui peuvent mimer l'aspect des lésions, une biopsie est habituellement réalisée pour conclure.
Traitement
En l'absence de symptôme, l'abstention thérapeutique est la règle. Si les lésions sont limitées, des traitements locaux, type dermocorticoïdes peuvent être essayés. En cas de forme étendue, des traitements systémiques (corticoïdes, acitrétine, voire biothérapie) ou la photothérapie ont leur place.
Iconographie
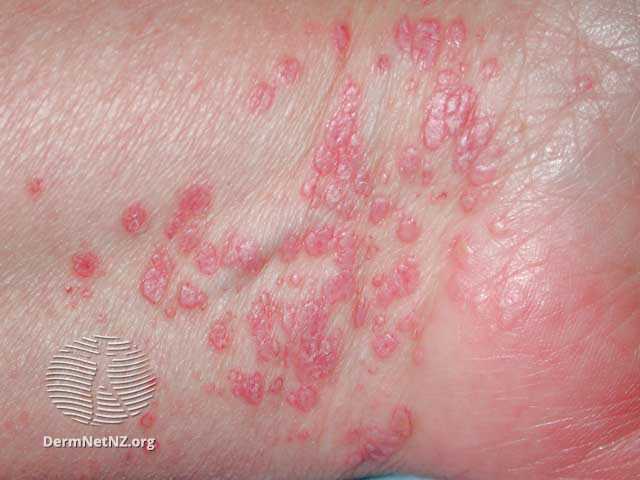

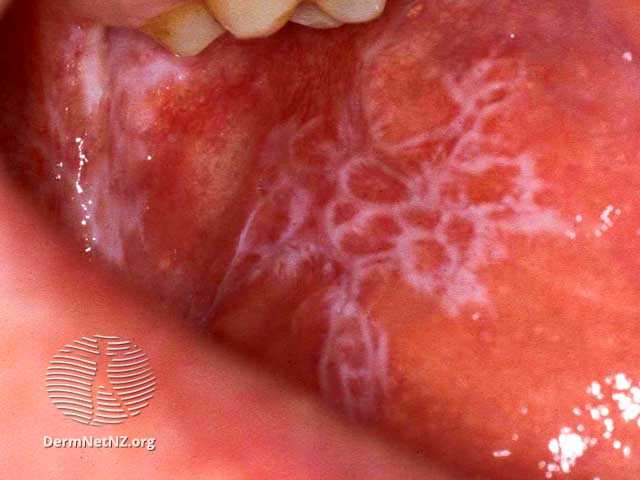
Items où il est abordé
Item 58 Sexualité normale et ses troubles
C'est une cause de dyspreunies d'intromission.
Item 116 Prurit
C'est une cause de prurit diffus dermatologique.
Item 155 Infections cutanéo-muqueuses et des phanères, bactériennes et mycosiques de l’adulte et de l’enfant
C'est un diagnostic différentiel de la candidose buccale, génitale, et unguéale.
Item 162 Infections sexuellement transmissibles (IST) : gonococcies, chlamydioses, syphilis, papillomavirus humain (HPV), trichomonose
C'est un diagnostic différentiel de la syphilis secondaire.
Item 168 Infections à herpès virus du sujet immunocompétent
C'est un diagnostic différentiel d'ulcérations buccales multiples.
Item 298 Tumeurs de la cavité buccale, naso-sinusiennes et du cavum, et des voies aérodigestives supérieures
C'est une lésion pré-cancéreuse des cancers de cavité buccale.
Item 302 Tumeurs cutanées, épithéliales et mélaniques
C'est une lésion pré-cancéreuse des carcinomes épidermoïdes cutanés.
Références
Pityriasis rosé de Gibert
En (très) bref
Épidémiologie
La fréquence du pityriasis rosé de Gibert n'est pas facile à trouver. Il touche des patient·e·s jeunes adultes ou enfants (mais pas les nourrissons). Le sex ratio est très légèrement en faveur de l'atteinte des femmes.
Signes
C'est une maladie dermatologique avec des lésions maculo-papuleuses rosées en forme de médaillon de 2 à 10 cm à bordure nette avec désquamation périphérique avec guérison centrale. On peut aussi voir des lésions plus petites de moins d'un centimètre moins bien limitées. Les lésions se localise surtout sur le tronc et la racine des membres. Elles évoluent sur moins de 3 mois sans traitement.
Causes
La cause précise de la maladie est inconnue. On suspecte une origine virale, peut-être en lien avec un des virus du groupe herpès.
Diagnostic/Examens complémentaires
Le diagnostic est le plus souvent clinique et ne nécessite pas d'examens complémentaires. Les lésions annulaires desquamantes à guérison centrale peuvent rendre le diagnostic difficile avec une dermatophytie de la peau glabre.
Traitement
Aucun traitement spécifique n'est nécessaire. En cas de prurit important, un traitement symptomatique peut être utile, via dermocorticoïdes par exemple.
Iconographie
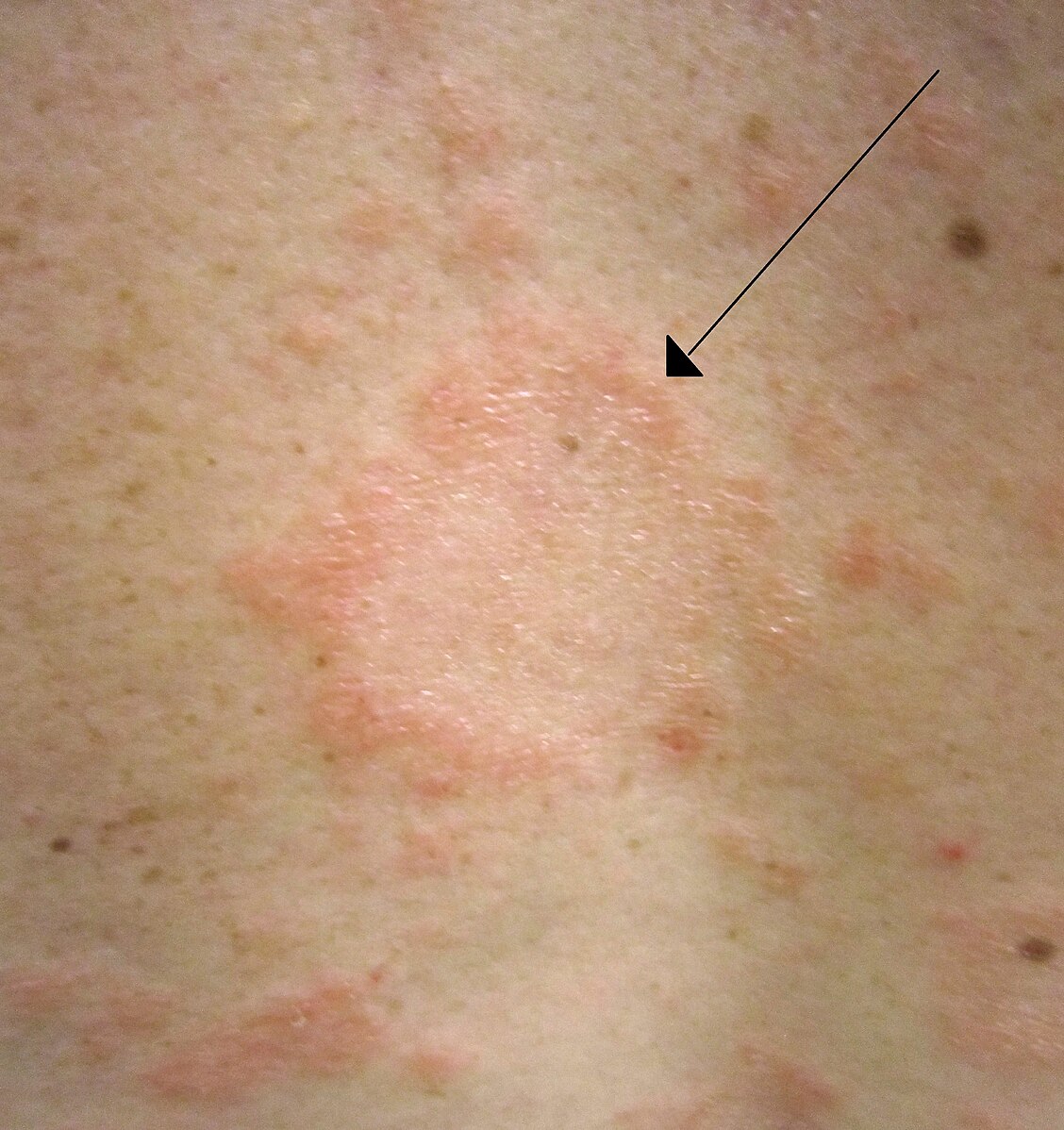
Items où il est abordé
Item 117 Psoriasis
C'est un diagnostic différentiel du psoriasis.
Références
Érythème polymorphe
En (très) bref
Épidémiologie
L'incidence précise est inconnue. L'âge moyen de survenue est entre 20 et 40 ans. Le sex ratio est légèrement en faveur de l'atteinte des hommes.
Signes
L'érythème polymorphe associe des lésions cutanées et muqueuses. Ces lésions apparaissent jusqu'à 15 jours après le facteur déclenchant. Elles se développent pendant environ 4 jours pour disparaître spontanément en environ 15 jours.
Signes cutanés
La lésion typique mesure généralement moins de 3 cm et se compose d'une macule ou une papule érythémateuse en cocarde constituée :
- d'une zone centrale qui peut être bulleuse ;
- un deuxième anneau, plus pâle ;
- un troisième anneau, erythémateux.
Les lésions se concentrent surtout sur les faces d'extension des extrémités, mais d'autres zones peuvent être atteintes.
Il n'y a généralement pas de prurit associé.
Signes muqueux
Les lésions muqueuses peuvent être un érythème, des érosions douloureuses, ou des bulles. Elles intéressent surtout les muqueuses oculaires, orales et génitales.
Causes
De très nombreuses situations peuvent être à l'origine de l'érythème polymorphe. Cela étant dit, les causes les plus fréquentes sont infectieuses avec l'infection à HSV et Mycoplasma pneumoniae.
Dans le cas de l'infection à HSV, les lésions seraient secondaires à une réaction immune dirigée contre des antigènes du virus présents dans la peau.
Par le passé, l'erythème polymorphe a été considéré comme une forme mineur du syndrome de Stevens-Johnson. Les deux entités sont aujourd'hui considérées comme distinctes.
Diagnostic/Examens complémentaires
Le diagnostic est clinique. Une biopsie cutanée peut être utile en cas de doute.
Traitement
Aucun traitement étiologique n'a fait la preuve de son efficacité. La prise en charge est donc généralement symptomatique et ambulatoire. Des mesures plus musclées sont parfois nécessaires, notamment en cas d'atteinte muqueuse sévère.
Iconographie
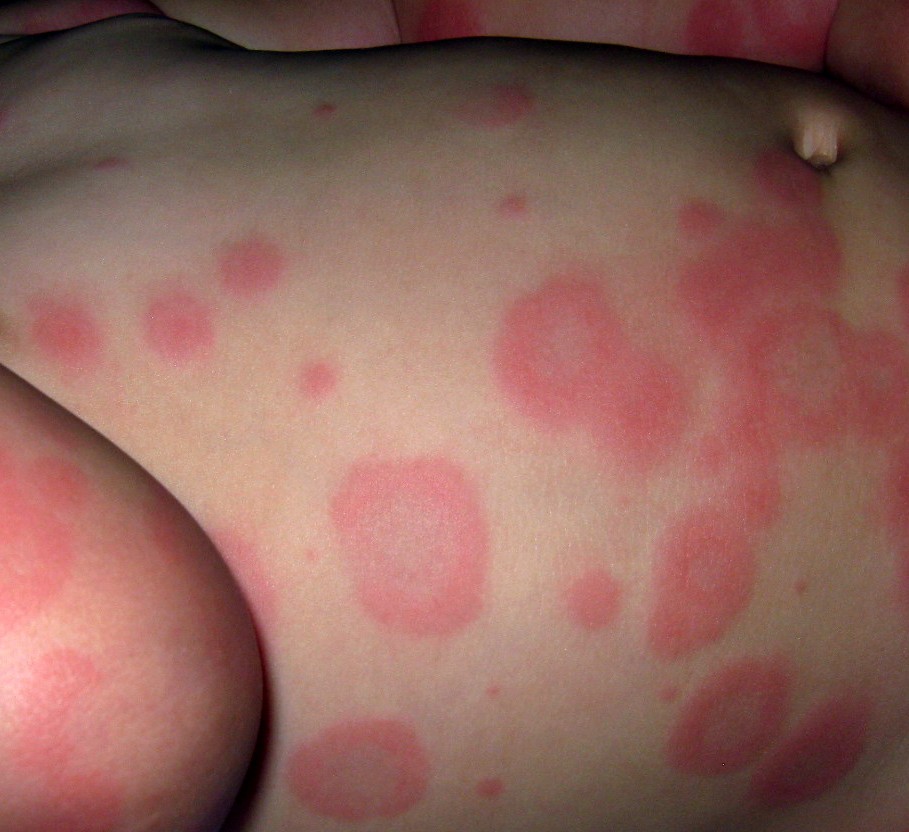
Items où il est abordé
Item 112 Dermatose bulleuse touchant la peau et/ou les muqueuses externes
C'est un diagnostic différentiel des dermatoses bulleuses auto-immunes.
Item 115 Toxidermies
C'est un diagnostic différentiel de la nécrolyse épidermique toxique.
Item 187 Hypersensibilités et allergies cutanéomuqueuses chez l’enfant et l’adulte. Urticaire, dermatites atopique et de contact
C'est un diagnostic différentiel de l'urticaire.
Références
Rhabdomyolyse
- En (très) bref
- Items où elle est abordée
- Item 97 Polyradiculonévrite aiguë
- Item 131 Trouble de la marche et de l'équilibre
- Item 166 Grippe
- Item 267 Troubles de l’équilibre acido-basique et désordres hydro-électrolytiques
- Item 330 Prescription et surveillance des classes de médicaments les plus courantes chez l’adulte et chez l’enfant, hors anti-infectieux
- Item 334 Polytraumatisé
- Item 337 Principales intoxications aiguës
- Item 342 Malaise, perte de connaissance, crise comitiale chez l'adulte
- Item 363 Fractures fréquentes de l'adulte et du sujet âgé
- Références
En (très) bref
Épidémiologie
Aux USA, on rapporte 26 000 cas chaque année, ce qui correspond à une incidence de 9 pour 100 000 habitants.
Signes
La rhabdomyolyse se définit par une destruction de cellules musculaires. Les signes sont en lien avec la perte de ces cellules et de leur membrane et traduisent le relargage du contenu cellulaire dans la circulation sanguine.
La triade classique associe :
- une douleur musculaire ;
- une faiblesse musculaire ;
- des urines foncées en lien avec une myoglobinurie.
D'autres signes cliniques peuvent s'observer :
- un œdème musculaire qui peut aller jusqu'à un véritable troisième secteur et au syndrome des loges ;
- de la fièvre.
Un certain nombre de perturbations biologiques, principalement hydro-électrolytiques sont présentes :
- tout d'abord des CPK très fortement augmentées au-delà de 15 000 UI/L ;
- une hyperkaliémie, une hyperphosphatémie et une hyperuricémie, en lien avec le relargage du contenu des myocytes ;
- une hypocalcémie consécutive à une chélation par les ions phosphates, qui fait place ensuite à une hypercalcémie, notamment en lien avec une libération du calcium intra-musculaire ;
- une acidose métabolique à trou anionique augmentée ;
- une insuffisance rénale aiguë en lien avec la myoglobinurie et la précipitation du phosphate et de l'acide urique.
Causes
On peut retenir 3 familles de causes :
- les atteintes musculaires traumatiques dont la station au sol prolongée ;
- les atteintes musculaires par exercice intense, qui peuvent survenir sur des myopathies pré-existantes ;
- les atteintes toxiques.
Diagnostic/Examens complémentaires
Le diagnostic repose sur l'association des signes cliniques et perturbations du bilan biologique sus-cités.
Traitement
La prise en charge associe :
- une expansion volémique ;
- une alcalinisation des urines ;
- le traitement de l'hyperkaliémie ;
- voire le recours à une expuration extra-rénale.
Items où elle est abordée
Item 97 Polyradiculonévrite aiguë
La rhabdomyolyse est un diagnostic différentiel du syndrome de Guillain-Barré.
Item 131 Trouble de la marche et de l'équilibre
La rhabdomyolyse est une complication de la station au sol prolongée.
Item 166 Grippe
La rhabdomyolyse est une complication non-respiratoire de la grippe.
Item 267 Troubles de l’équilibre acido-basique et désordres hydro-électrolytiques
L'hypokaliémie peut se compliquer de rhabdomyolyse.
Item 330 Prescription et surveillance des classes de médicaments les plus courantes chez l’adulte et chez l’enfant, hors anti-infectieux
La rhabdomyolyse est un effet indésirable de plusieurs hypolipémiants dont les statines.
Item 334 Polytraumatisé
La rhabdomyolyse est une complication des traumatismes des membres.
Item 337 Principales intoxications aiguës
L'intoxication au monoxyde de carbone peut induire une rhabdomyolyse.
Le toxidrome sérotoninergique peut inclure une rhabdomyolyse.
Item 342 Malaise, perte de connaissance, crise comitiale chez l'adulte
La rhabdomyolyse est une complication de l'état de mal épileptique.
Item 363 Fractures fréquentes de l'adulte et du sujet âgé
La rhabdomyolyse est une complication des traumatismes des membres.
Références
Glomérulonéphrite membrano-proliférative
En (très) bref
Épidémiologie
La prévalence des formes primitives est de l'ordre de 20 pour 100 0001. Les formes primitives concernent l'enfant et l'adulte entre 8 et 30 ans principalement. Les formes secondaires concernent plutôt les adultes après 30 ans. Le sex ratio est équilibré.
Signes
La définition de la glomérulonéphrite membrano-proliférative est une entité histologique qui correspond à la présence :
- d'une hypercellularité mésangiale ;
- d'une prolifération endocapillaire ;
- d'un épaississement de la membrane basale glomérulaire, avec un aspect de double contour.
Cliniquement, on retrouve un syndrome glomérulaire. Il peut, comme il ne peut pas, y avoir une insuffisance rénale. Au sédiment urinaire, on trouve une protéinurie variable, une hématurie, des hématies dysmorphique caractéristique et parfois des rouleaux.
Causes
En fonction des résultats de l'immunofluorescence, les glomérulonéphrites membrano-prolifératives peuvent être subdivisées en 3 catégories qui ont chacune leurs causes.
Médiée par des immunoglobulines ou des complexes immuns
Sous cette catégorie, on rassemble des maladies auto-immunes (dont le syndrome de Gougerot-Sjögren), des infections chroniques (dont l'hépatite C), et des gammapathies monoclonales.
Médiée par le complément
Elles sont en lien avec des maladies rares du complément.
Sans dépôt d'immunoglobulines ou de complément
Parmi les causes les plus au programme, on trouve les séquelles de microangiopathie thrombotique, le syndrome des anti-phospholipides, les néphrites radiques, et l'hypertension maligne.
Diagnostic/Examens complémentaires
Le diagnostic est posé sur la biopsie rénale. Le dosage du complément est également utile.
Traitement
Dans les formes secondaires, le traitement est celui de la cause. Un traitement néphroprotecteur par inhibiteur du SRAA est souvent utilisé.
Dans les formes primitives de l'enfant, en présence d'un syndrome néphrotique, les corticoïdes ont parfois leur place.
Items où elle est abordée
Item 167 Hépatites virales
C'est une manifestation extra-hépatique de l'hépatite C.
Item 260 Hématurie
C'est une cause d'hématurie néphrologique.
Item 261 Néphropathies glomérulaires
C'est un diagnostic différentiel de la glomérulonéphrite aiguë post-infectieuse.
Références
Brucellose
En (très) bref
Épidémiologie
Dans les pays dit développés dont la France, la maladie est éradiquée et les cas sont tous d'importation. La maladie est surtout présente au Moyen-Orient et sur la côte méditerranéenne sud.
Signes
La maladie peut évoluer en plusieurs phases dont les plus en lien avec le programme sont les suivantes.
Phase aiguë
Les signes de la brucellose aiguë sont :
- fièvre et sueurs ondulant sur 3-4 périodes d'une quinzaine de jours ;
- un syndrome tumoral avec hépato-spléno-mégalie et adénopathie ;
- des arthro-myalgies
Phase secondaire focalisée
Les signes de la brucellose secondaire focalisée sont :
- une spondylodiscite, ou d'autres localisations articulaires (sacro-iliite, arthrite de hanche, ostéites) ;
- des signes neurologiques : méningite à liquide clair hypoglycorachique, méningo-encéphalité, méningo-myélo-radiculite ;
- une endocardite, plutôt rare mais mentionnée dans le référentiel de médecine cardio-vasculaire.
Causes
La brucellose est une zoonose en lien avec des bactéries du genre Brucella. Le réservoir est animal uniquement. La contamination se fait au contact de bovins, ovins, porcs ou chiens, que ce soit par contact direct avec leur placenta et sécrétions génitales, par l'alimentation, voire par voie aérienne.
Diagnostic/Examens complémentaires
Le diagnostic est clinico-biologique et repose donc sur des hémocultures, la sérologie, ou la PCR.
Traitement
Le traitement est une bi-antibiothérapie associant une tétracycline d'une part, et de la rifampicine ou un aminoside d'autre part. Pour les formes aiguës, il dure 6 semaines. Dans les formes secondaires, il ne doit pas durer moins de 3 mois.
Items où elle est abordée
Item 152 Endocardite infectieuse
C'est une cause d'endocardite à germes à croissance difficile.
Item 156 Infections ostéo-articulaires
C'est une cause rare de spondylodiscite.
Item 211 Sarcoïdose
C'est une cause infectieuse de granulome.
Item 212 Hémogramme
C'est une cause d'infection bactérienne sans hyperleucocytose.
Item 217 Syndrome mononucléosique
C'est une cause rare de syndrome mononucléosique.
Item 220 Adénopathies superficielles
C'est une cause infectieuse de polyadénoapthie.
Item 296 Agranulocytose médicamenteuse
C'est une cause infectieuse de neutropénie.
Références
Rickettsioses
En (très) bref
- le groupe boutonneux qui inclue notamment la fièvre boutonneuse méditerranéenne ;
- les groupe typhus qui inclue notamment le typhus épidémique.
Fièvre boutonneuse méditerranéenne
Épidémiologie
Comme son nom l'indique, on la retrouve dans le sud de l'Europe et le pourtour méditéranéen. La majorité des cas sont observés l'été.
Signes
Après une incubation d'environ 7 jours, apparaît brutalement la triade suivante :
- fièvre en plateau ;
- exanthème maculo-papuleux, atteignant les paumes et les plantes ;
- escarre d'inoculation au niveau de la piqûre de tique.
D'autres signes moins spécifiques (céphalée, arthralgie, adénopathie) peuvent être présents.
Cependant la maladie est asymptomatique dans 50% des cas et c'est une maladie bénigne dans l'extrême majorité des autres cas.
Causes
La bactérie en cause est Rickettsia conorii inoculée par des tiques de chien.
Diagnostic/Examens complémentaires
Le diagnostic clinique suffit pour commencer le traitement. Il sera cependant confirmé par une sérologie. D'autres techniques (PCR ou culture) sont réservées aux centres disposant d'un plateau technique suffisant.
Traitement
Le traitement de référence est la doxycycline en dose unique. On la commence dès que les prélèvements diagnostiques ont été faits.
Iconographie
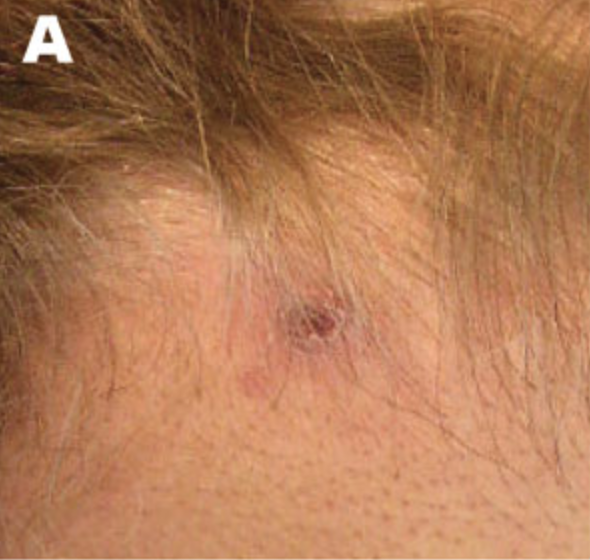
Typhus épidémique
Épidémiologie
Le typhus épidémique se rencontre surtout en Amérique centrale, en Amérique du sud, en Afrique et en Asie.
Signes
Après une incubation d'environ 12 jours, apparaissent brutalement fièvre, céphalée et myalgie. D'autres symptômes non spécifiques (troubles gastro-intestinaux, arthralgie) peuvent être présents. Des atteintes d'organes sont possibles :
- pulmonaire avec bronchite, pneumopathie interstitielle, bronchiolite ;
- splénomégalie ;
- conjonctivite.
Les complications cardiaques et neurologiques rendent la maladie potentiellement grave.
Causes
La bactérie en cause est Rickettsia prowazekii. Elle est transmise par Pediculus humanis corporis.
Diagnostic/Examens complémentaires
Le diagnostic clinique suffit pour commencer le traitement. Il sera cependant confirmé par une sérologie. D'autres techniques (PCR ou culture) sont réservés aux centres disposant d'un plateau technique suffisant.
Traitement
Le traitement de référence est la doxycycline en dose unique. On le commence dès que les prélèvements diagnostiques ont été faits.
Items où elles sont abordées
Item 83 Inflammation occulaire
Les rickettsioses sont une cause infectieuse d'uvéite postérieure.
Item 171 Gale et pédiculose
Le typhus épidémique est transmis par Pediculus humanis corporis.
Item 175 Voyage en pays tropical
C'est une cause de fièvre au retour d'un pays tropical avec une incubation comprise entre 7 et 14 jours.
Item 217 Syndrome mononucléosique
C'est une causes rare de syndrome mononucléosique.
Références
Histoplasmose
En (très) bref
Épidémiologie
Les zones les plus atteintes par l'histoplasmose sont l'Amérique du Sud et l'Amérique centrale, l'Afrique du Sud, l'Asie du Sud-Est, et la Nouvelle-Zélande. La maladie touche préférentiellement les hommes avec un sex ratio de 4 hommes pour 1 femme.
Signes
Il existe plusieurs formes de la maladie selon le profil de la personne infectée.
Histoplasmose pulmonaire aiguë
Cette forme touche les personnes immunocompétentes indemnes de maladie pulmonaire.
Les signes sont ceux d'une infection respiratoire basse non spécifique, auxquels peuvent s'associer des arthralgies, un érythème noueux, un érythème polymorphe, voire un péricardite.
La maladie guérit d'elle-même en une dizaine de jours.
Histoplasmose pulmonaire chronique
Cette forme touche les personnes immunocompétentes avec poumon pathologique.
Les signes pulmonaires sous-jacents sont exacerbés. On peut voir en plus des sueurs nocturnes, des douleurs thoraciques et une hémoptysie. L'évolution de la maladie va vers l'apparition de lésions cavitaires.
Histoplasmose disséminée
Cette forme touche les personnes immunodéprimées.
Les signes peuvent atteindre tous les organes ou presque : signe respiratoire, cutané, syndrome tumoral, méningo-encéphalite, cytopénie, état de choc, etc.
Causes
L'histoplasmose est due à un champignon Histoplasma capsulatum. Le variant traité dans ce chapitre est capsulatum. On le trouve dans les sols acides riche en déjections d'oiseaux et chauve-souris.
La contamination se fait par voie aérienne par inhalation.
Diagnostic/Examens complémentaires
Le champignon peut être mis en évidence sur de nombreux prélèvements. Le lavage broncho-alvéolaire est le plus rentable. La sérologie a également sa place.
Traitement
Le traitement n'est pas systématique dans les formes pulmonaires (aiguës ou chroniques). Le choix dépendra des symptômes présents. En revanche, il est systématique dans les formes disséminées. Le traitement repose alors sur l'itraconazole ou l'amphotéricine B.
Items où elle est abordée
Item 207 Opacités et masses intra-thoraciques chez l’enfant et chez l’adulte
C'est une cause d'adénopathie du médiastin moyen non tumorale.
Item 211 Sarcoïdose
C'est une cause infectieuse de granulome.
Références
Glaucome néo-vasculaire
En (très) bref
Épidémiologie
Le glaucome néo-vasculaire représenterait autour de 5% des causes de glaucome. Le sex ratio et l'âge moyen au diagnostic sont le reflet des causes de la maladie.
Signes
Avant que le glaucome se développe, on observera une rubéose irienne qui reflète la formation des néo-vaisseaux.
L'apparition du glaucome peut se faire plus ou moins rapidement. On parle parfois de la règle des 100 jours pour traduire l'apparition d'un glaucome 100 jours après une occlusion de la veine centrale de la rétine ischémique.
Causes
C'est un glaucome due à une fermeture de l'angle irido-cornéen par des néo-vaisseaux. Leur prolifération est majoritairement initiée par une ischémie rétinienne. Une trame fibro-vasculaire se forme à la périphérie de l'iris et au niveau de l'angle. Son évolution finit par obstruer le circuit de drainage de l'humeur aqueuse, contribuant à augmenter la pression intra-oculaire, provoquant à terme un glaucome.
On retient 3 principales causes :
- l'occlusion de la veine centrale de la rétine ischémique ;
- la rétinopathie diabétique proliférante ;
- une anomalie carotidienne compromettant la vascularisation de l'œil.
À noter que les occlusions de l'artère centrale de la rétine ne seraient pas assez complètes pour induire une ischémie rétinienne.
Diagnostic/Examens complémentaires
Le diagnostic repose sur l'anamnèse, l'examen oculaire aidé de la gonioscopie, et la mise en évidence d'une rubéose irienne.
Traitement
La prise en charge s'articule sur 2 plans :
- corriger l'ischémie rétinienne via la panphotogocagulation rétinienne et l'injection intra-vitréenne d'anti-VEGF ;
- diminuer la pression oculaire en utilisant les mêmes principes que pour le glaucome primitif à angle ouvert.
Iconographie
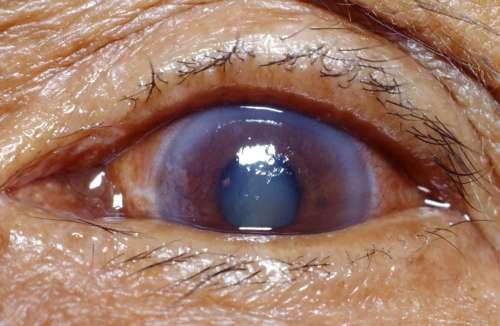
Items où il est abordé
Item 82 Altération aiguë de la vision
C'est une complication de l'occlusion de la veine centrale de la rétine.
Item 84 Glaucome
C'est une cause de glaucome à angle fermé.
Item 247 Diabète
C'est une complication de la rétinopathie diabétique proliférante.
Références
Ostéonécrose aseptique
En (très) bref
Épidémiologie
L'incidence exacte n'est pas connue, mais aux États-Unis elle serait d'environ 5 pour 100 000 habitants. Le sex-ratio dépend de la cause sous-jacente.
Signes
Le symptôme clé est la douleur dont la localisation dépendra de l'os atteint.
On peut aussi trouver un épanchement articulaire mécanique.
Causes
Le processus histologique est une réduction de la vascularisation qui conduit à un infarctus osseux en plusieurs mois. La physiopathologie exacte n'est pas encore élucidée.
Les causes sont variées, en voici quelques unes.
Fracture
C'est la cause en tête de liste.
Médicaments et toxiques
- Corticoïdes
- Biphosphonates
- Radiothérapie
- Alcool
- Tabac
Maladies systémiques
- Lupus érythémateux disséminé
- Drépanocytose
- Maladie de Gaucher
Diagnostic/Examens complémentaires
Le diagnostic se fait sur un examen d'imagerie.
Radiographie
Normale au début, les signes se voient après plusieurs mois :
- changement de densité ;
- puis sclérose.
Dans l'ostéonécrose de la tête fémorale, cela se traduit par une perte de la sphéricité.
IRM
C'est l'examen de référence.
Traitement
Les plus petites lésions peuvent ne pas être traitées. Autrement la chirurgie s'impose.
Iconographie
Items où elle est abordée
Item 23 Maladies rares
C'est une complication des crises vaso-oclusives de la drépanocytose.
Item 128 Ostéopathies fragilisantes
L'ostéonécrose de la mâchoire est un effet indésirable des biphosphonates.
Item 129 Arthrose
C'est un diagnostic différentiel de l'arthrose.
Item 194 Lupus systémique. Syndrome des anti-phospholipides
C'est un signe du lupus érythémateux disséminé.
Item 199 Syndrome douloureux régional complexe
C'est un diagnostic différentiel du SDRC.
Item 200 Douleur et épanchement articulaire
C'est une cause d'épanchement articulaire mécanique sans anomalie à la radiographie.
Item 363 Fractures fréquentes de l'adulte et du sujet âgé
C'est une complication des fractures de l'extrémité supérieure du fémur, principalement les Garden 3 et 4.
Références
Fausse couche
- En (très) bref
- Items où elle est abordée
- Item 24 Principales complications de la grossesse
- Item 25 Grossesse extra-utérine
- Item 27 Prévention des risques fœtaux : infection, médicaments, toxiques, irradiation
- Item 29 Connaître les principaux risques professionnels pour la maternité, liés au travail de la mère
- Item 40 Algies pelviennes chez la femme
- Item 44 Tuméfactions pelviennes chez la femme
- Item 45 Spécificités des maladies génétiques
- Item 173 Zoonoses
- Item 194 Lupus systémique. Syndrome des anti-phospholipides
- Item 255 Diabète gestationnel
- Références
En (très) bref
Épidémiologie
La fausse couche est quelque chose de très courant puisqu'elle concerne 10% des grossesse connues et jusqu'à 30% si l'on inclue les grossesses non détectées. Elle a majoritairement lieu au premier trimestre. Les fausses couches du second trimestre touche moins de 1% des grossesses.
Signes
Une fausse couche est définie en France par l'expulsion spontanée du contenu utérin avant 22 SA. On parle de fausse couche précoce avant 14 SA (pendant le 1er trimestre donc) et tardive ensuite.
La présentation clinique la plus fréquente associe un saignement génital et des douleurs pelviennes. Généralement non compliquée, il peut néanmoins y avoir une syndrome hémorragique ou une infection type chorio-amniotite.
Causes
Deux tiers des fausses couches sont en lien avec des anomalies chromosomiques de l'embryon ou du fœtus, qu'ils s'agissent d'anomalies de nombre ou de structure.
Des anomalies anatomiques maternelles gênant l'implantation et le développement de l'embryon ou du fœtus peuvent aussi être en cause : fibromes, polypes, synéchies, etc.
Au deuxième trimestre, les causes sont beaucoup plus variées.
Diagnostic/Examens complémentaires
Le diagnostic repose sur l'échographie pelvienne et le dosage de β-hCG.
Traitement
Le traitement (au programme) peut être l'abstention thérapeutique, ou bien un traitement médical par prostaglandine ou l'aspiration endo-utérine selon que la fausse couche est complète ou non.
Items où elle est abordée
Item 24 Principales complications de la grossesse
C'est une cause d'hémorragie au premier trimestre de grossesse.
Item 25 Grossesse extra-utérine
C'est un diagnostic différentiel des grossesses extra-utérines.
Item 27 Prévention des risques fœtaux : infection, médicaments, toxiques, irradiation
La listeriose, la rougeole, la consommation d'opiacés, de cocaïne ou de tabac sont des facteurs de risque de fausse couche spontanée.
Item 29 Connaître les principaux risques professionnels pour la maternité, liés au travail de la mère
Le travail posté et le travail de nuit après 12 SA sont des facteurs de risque de fausse couche spontanée.
Item 40 Algies pelviennes chez la femme
C'est une cause obstétricale de douleurs pelviennes aiguës.
Item 44 Tuméfactions pelviennes chez la femme
Les fibromes sont des facteurs de risque de fausse couche spontanée.
Item 45 Spécificités des maladies génétiques
C'est une complication des prélèvements pré-nataux pour le diagnostic de la trisomie 21.
Item 173 Zoonoses
La fièvre Q est une facteur de risque de fausse couche spontanée.
Item 194 Lupus systémique. Syndrome des anti-phospholipides
Les fausses couches spontanés répétées font partie des critères de classification du syndrome des anti-phospholipides.
Item 255 Diabète gestationnel
Le diabète gestationnel est un facteur de risque de fausse couche spontanée.
Références
Conclusion
Et voilà ! Vous en avez fini avec les items fantômes que nous avions repérés. Si vous en identifiez d'autres, n'hésitez pas à nous en faire part à l'adresse mentionnée dans l'introduction.
Take-home message
Gardez bien en tête que le concours de l'internat porte avant tout sur les pathologies fréquentes ou graves, idéalement les deux. Il est donc peu probable qu'apprendre les éléments développés dans cet ouvrage soit plus rentable que travailler vos référentiels. Les différents chapitres que vous avez parcourus ont davantage pour but d'élargir votre culture médicale et lever légèrement le voile de termes opaques omniprésents.
Vers l'infini et l'au-delà
Comme mentionné dans l'introduction, il existe sûrement des items fantômes qui ne sont pas encore répertoriés dans cet ouvrage. Nous avons déjà quelques idées pour compléter le Pokédex, mais n'hésitez pas à nous faire des propositions via l'adresse contact.
Si vous avez identifié des erreurs lors de votre lecture, n'hésitez pas à nous les faire remonter (toujours à l'adresse contact). Le format numérique permet une mise à jour rapide.
Enfin si vous voulez découvrir d'autres contenus satellites du deuxième cycle des études médicales, vous pouvez vous rendre sur mon site internet !
Bonus
Comment accéder à UpToDate ?
UpToDate est un site de recommandations américains qui est une petite mine d'or d'informations sourcées par des publications scientifiques. C'est une des sources sur lesquelles se basent certains chapitres.
Si vous êtes étudiant·e hospitalier·ère à l'AP-HP, vous pouvez y accéder depuis la page d'accueil de l'intranet AP-HP.
- Rendez-vous dans Afficher toutes les applications
- Rechercher UpToDate que vous devriez trouver sous un des hôpitaux de Sorbonne Universités, par exemple Saint-Antoine
- Vous pouvez ainsi vous créer un compte auquel vous pourrez accéder depuis chez vous !
Si vous êtes dans une UFR non parisienne, il est possible que votre CHU y soit abonné et qu'une méthode similaire vous permettent d'y accéder. Vous pouvez également essayer de vous rendre sur leur site en étant connecté au réseau de votre université, si les bons abonnements sont là, vous pourrez y accéder sans connexion (testé et approuvé à Limoges).
Comment trouver les Protocoles Nationaux de Diagnostic et de Soins ?
Rien de bien compliqué à première vue : il suffit d'une recherche Google. Ceci étant dit, les filières maladie rare qui éditent ces plans nationaux de diagnostic et de soins produisent parfois d'autres documents pour aider à mieux comprendre ces pathologies. Une fois la filière identifiée via la page de garde du PNDS, rendez-vous sur leur site pour y atteindre la page de la maladie : il y a parfois de bonnes surprises !
Comment accéder à des publications scientifiques ?
Nous faisons parfois référence à des articles scientifiques. Un tutoriel détaillé sur l'accès à la littérature scientifique dépasse de très loin la portée de cet ouvrage. Voici néanmoins quelques outils qui vous permettrons de vous lancer :
- le catalogue en ligne du service commun de la documentation de votre université (le site de votre BU) ;
- Google Scholar.